ketikan dari hal. 399 - 409.docx
TRANSCRIPT

GABAA Receptor Subtypes
GABAA receptors play a critical role in mediating inhibitory neurotransmission
and as targets of the anxiolytic benzodiazepines. The molecular structure of
GABAA receptors is shown in figure 9-21. Each subunit of a GABAA receptor has
four transmembrane regions (Figure 9-21A). When five subunits cluster together,
they form an intact GABAA receptor with a chloride channel in the center (Figure
9-21B). There are many different subtypes of GABAA receptors, depending upon
which subunits are present (Figure 9-21C). Subunits of GABAA receptors are
sometimes also called isoforms, and include a (with six isoforms, α1 to α6), β (with
three isoforms, β1 to β3) γ (with three isoforms, γ1 to γ3), δ, ε, π, θ, dan ρ (with
three isoforms, ρ1 to ρ3) (Figure 9-21C). What is important for this discussion is
that, depending upon which subunits are present, the functions of a GABAA
receptor can vary significantly.
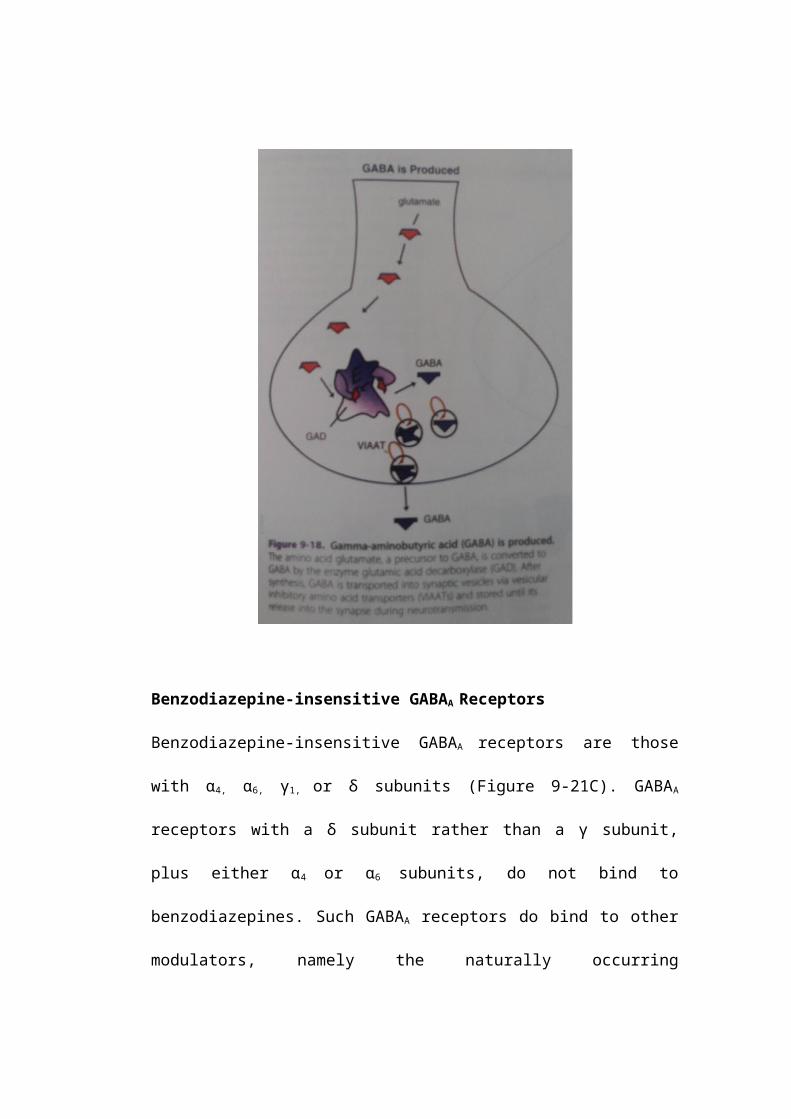
Benzodiazepine-insensitive GABAA Receptors
Benzodiazepine-insensitive GABAA receptors are those with α4, α6, γ1, or δ subunits
(Figure 9-21C). GABAA receptors with a δ subunit rather than a γ subunit, plus
either α4 or α6 subunits, do not bind to benzodiazepines. Such GABAA receptors do
bind to other modulators, namely the naturally occurring neurosteroids, as well as
to alcohol and to some general anesthetics (Figure 9-21C). The binding site for
these non-benzodiazepine modulators is located between the α and the δ subunits,
one site per receptor complex (Figure 9-21C). Two molecules of GABA bind per
receptor complex, at sites located between the α and the β subunits, sometimes

referred to as the GABA agonist site (Figure 9-21C). Since the site for the
modulators is in a different location from the agonist sites for GABA, the
modulatory site is often called allosteric (literally, “other site”), and the agents
that bind there are called allosteric modulators.
Benzodiazepine-insensitive GABAA receptor subtypes (with δ subunits)
are located extrasynaptically, where they capture not only GABA that diffuses
away from the synapse, but also neurosteroids synthesized and released by glia
(Figure 9-22). Extrasynaptic, benzodiazepine-insensitive GABAA receptors are
thought to mediate a type of inhibition at the postsynaptic neuron that is tonic, in
contrast to the phasic type of inhibition mediated by postsynaptic benzodiazepine-
sensitive GABAA receptors (Figure 9-22). Thus, tonic inhibition may be regulated
by the ambient levels of extracellular GABA molecules that have escaped
presynaptic reuptake and enzymatic destruction. Tonic inhibition is thought to set
the overall tone and excitability of the postsynaptic neuron, and to be important

for certain regulatory events such as the frequency of neuronal discharge in
response to excitatory inputs.
Since the GABAA receptors that modulate this action are not sensitive
to benzodiazepines, they are not likely to be involved in the anxiolytic actions of
benzodiazepines in various anxiety disorders. However, novel hypnotics as well
as anesthetics have targeted these extrasynaptic benzodiazepine-insensitive
GABAA receptors, and it is possible that novel synthetic neurosteroids that also
target benzodiazepine-insensitive GABAA receptor subtypes could some day
become novel anxiolytics. Indeed, anxiety itself may in part be dependent upon
having the right amount of tonic inhibition in key anatomic areas such as the
amygdala and cortical areas of CSTC loops. Furthermore, naturally occurring
neurosteroids may be important in setting that inhibitory tone in critical brain
areas. If this tone becomes dysregulated, it is possible that abnormal neuronal
excitability could become a factor in the development of various anxiety
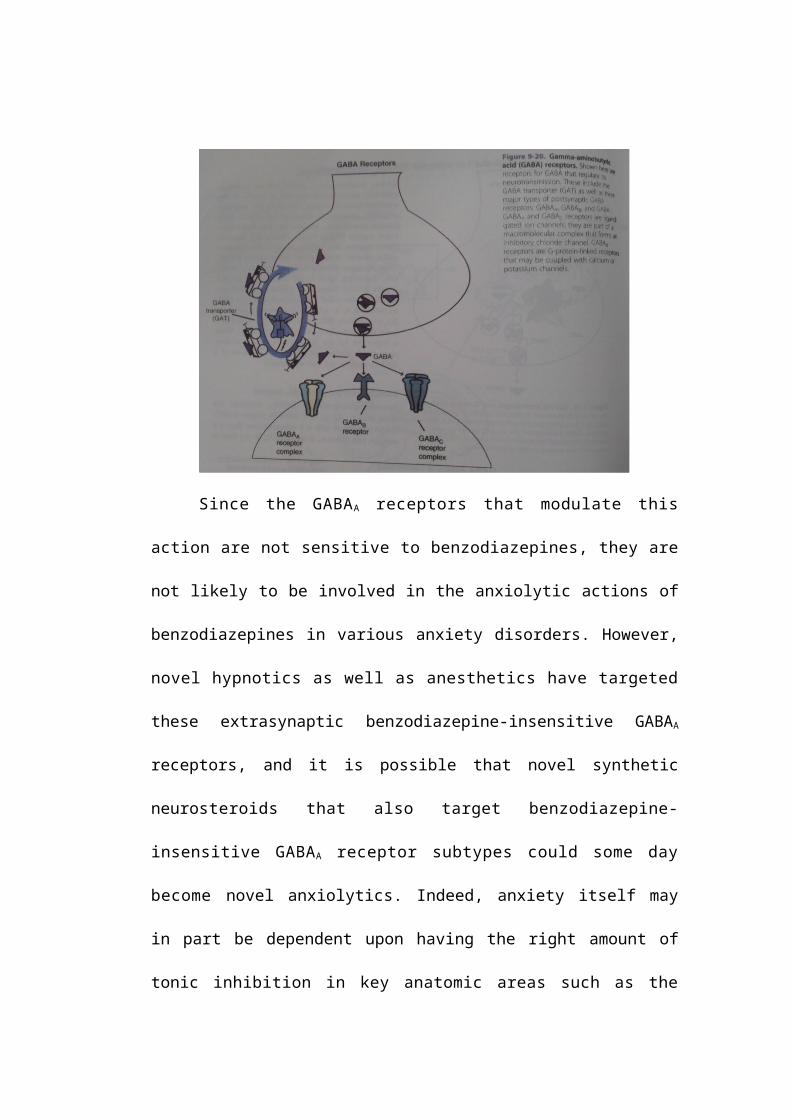
disorders.

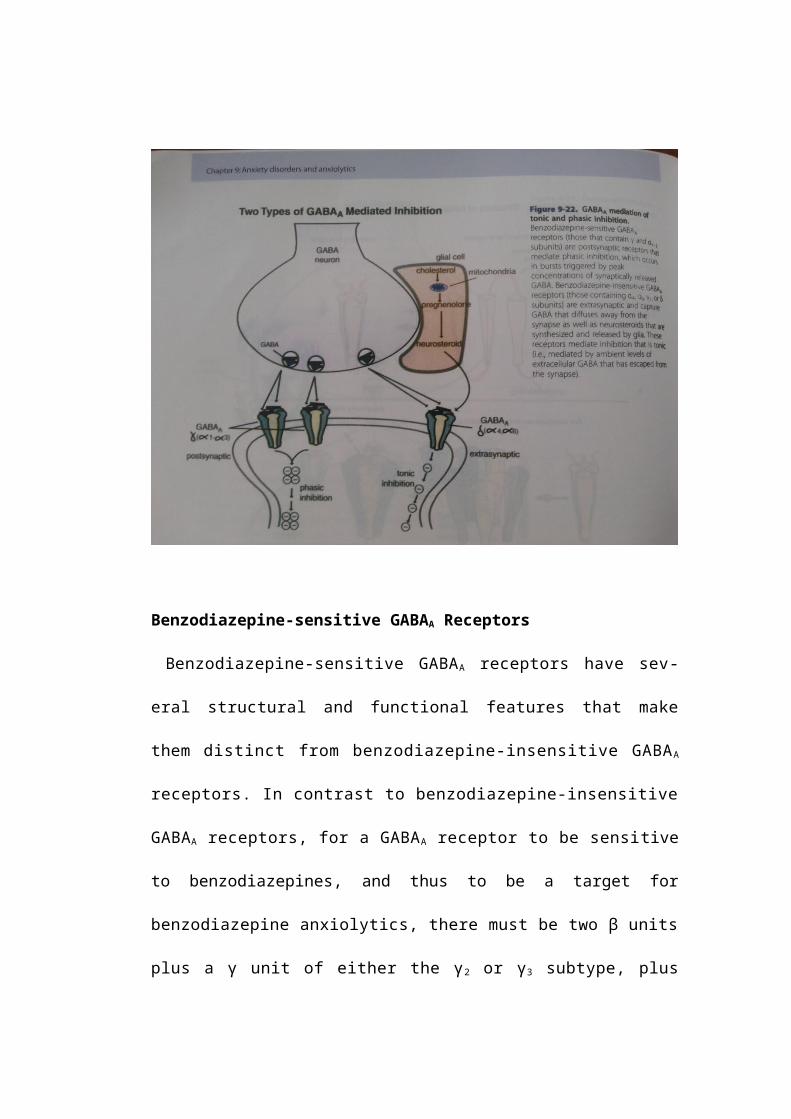
Benzodiazepine-sensitive GABAA Receptors
Benzodiazepine-sensitive GABAA receptors have several structural and
functional features that make them distinct from benzodiazepine-insensitive
GABAA receptors. In contrast to benzodiazepine-insensitive GABAA
receptors, for a GABAA receptor to be sensitive to benzodiazepines, and thus to
be a target for benzodiazepine anxiolytics, there must be two β units plus a γ
unit of either the γ2 or γ3 subtype, plus two α units of the α1, α2, or α3,
subtype (Figure 9-21C). Benzodiazepines appear to bind to the region of
the receptor between the γ2/3 subunit and the α1/2/3 subunit, one benzodiazepine
molecule per receptor complex (Figure 9-21C). GABA itself binds with two
molecules of GABA per receptor complex to the GABA agonist sites in the

regions of the receptor between the α and the β units (Figure 9-21C).
Benzodiazepine-sensitive GABAA receptor subtypes (with γ subunits and α I-3
subunits) are thought to be postsynaptic in location and to mediate a type of
inhibition at the postsynaptic neuron that is phasic, occurring in bursts of
inhibition that are triggered by peak concentrations of synaptically released
GABA (Figure 9-22). Theoretically, benzodiazepines acting at these receptors,
particularly the α2/3 subtypes clustered postsynaptic GABA sites, should exert
an anxiolytic effect due to enhancement of phasic postsynaptic inhibition. If
this action occurs at overly active output neurons in the amygdala or in
CSTC loops, it would theoretically cause anxiolytic actions with reduction of
both fear and worry.
Not all benzodiazepine-sensitive GABAA receptors are the same. Notably, those
benzodiazepine-sensitive GABAA receptors with α l subunits may be most
important for regulating sleep and are the presumed targets of numerous
sedative-hypnotic agents, including both benzodiazepine and non-
benzodiazep positive allosteric modulators of the GABAA, receptor (Figure 9-
21C). The α1 subtype of GABAA receptors and the drugs that bind to it are
discussed further in Chapter 11 on sleep. Some of these agents are selective for
only the α1 subtype of GABAA receptor.
On the other hand, benzodiazepine-sensitive GABAA receptors with α2
(and/or α3) subunits may be most important for regulating anxiety and are the
presumed targets of the anxiolytic benzodiazepines (Figure 9-21C). However,
currently available benzodiazepines are nonselective for GABAA receptors with
different α subunits. Thus, there is an ongoing search for selective α2/3 agents that
could be utilized to treat anxiety disorders in humans. Such agents would theor-
etically be anxiolytic without being sedating. Partial agonists selective for α2/3
subunits of benzodiazepine-sensitive GABAA receptors hypothetically would
cause less euphoria, be less reinforcing and thus less abusable, cause less
dependence, and cause fewer problems in withdrawal. Such agents are being
investigated but have not yet been introduced into clinical practice. Abnormally
expressed γ2, α2, or δ subunits have all been associated with different types of
epilepsy. Receptor subtype expression can change in response to chronic
benzodiazepine administration and withdrawal, and could theoretically be
altered in patients with various anxiety-disorder subtypes.
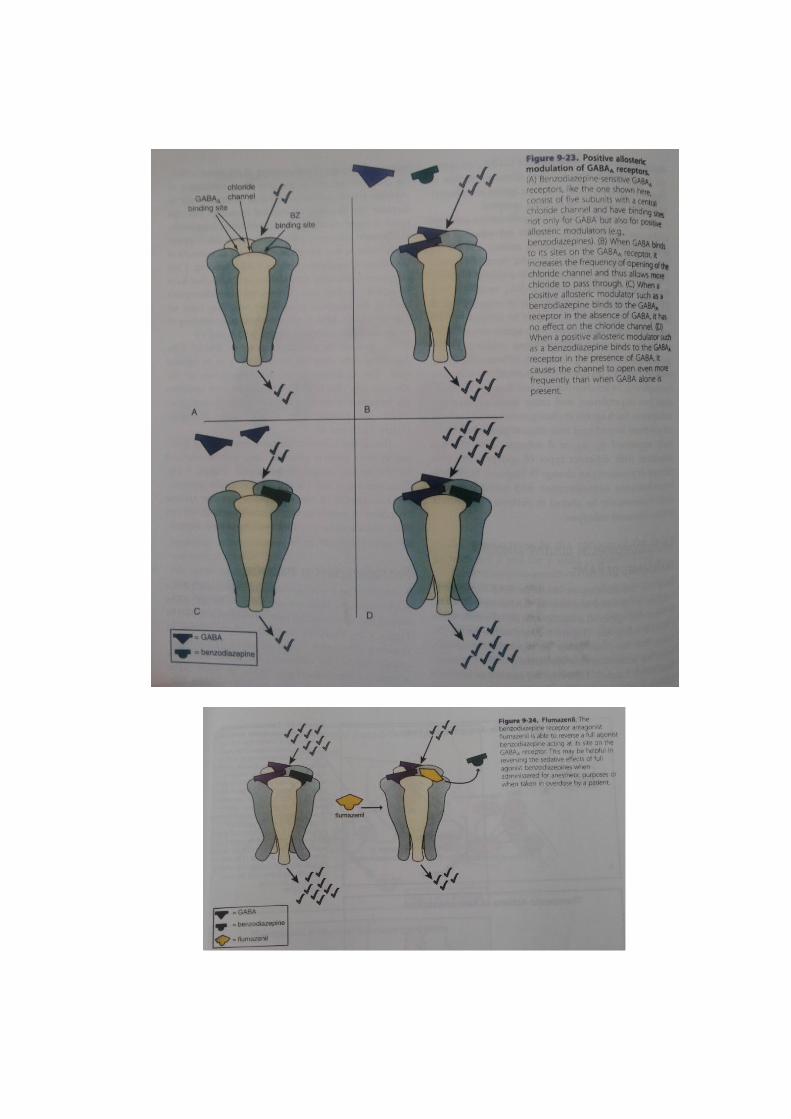
Benzodiazepines as positive allosteric modulators or PAMs
Since the benzodiazepine-sensitive GABAA receptor complex is regulated not
only by GABA itself, but also by benzodiazepines at a highly specific allosteric
modulator binding site (Figure 9-23), this has led to the notion that there may
be an "endogenous" or naturally occurring benzodiazepine synthesized in the
brain (the brain's own Xanax!). However, the identity of any such substance
remains elusive. Furthermore, it is now known that synthetic drugs that do not
have a benzodiazepine structure also bind to the "benzodiazepine receptor."
These developments have led to endless confusion with terminology, since non-
benzodiazepines also bind to the "benzodiazepine receptor!" Thus, many experts
now call the "benzodiazepine site" the GABAA allosteric modulatory site and

anything that binds to this site, including benzodiazepines, allosteric modulators.
Acting alone, GABA can increase the frequency of opening of the chloride
channel, but only to a limited extent (compare Figure 9-23A and B). The
combination of GABA with benzodiazepines is thought to increase the frequency
of opening of inhibitory chloride channels but not to increase the conductance
of chloride across individual chloride channels, nor to increase the duration
of channel opening. The end result is more inhibition. More inhibition
supposedly yields more anxiolytic action. How does this happen? The answer
is that benzodiazepines act as agonists at the allosteric modulatory site of
GABA binding. They are positive allosteric modulators, or PAMs, but have no
activity on their own. Thus, when benzodiazepines bind to the allosteric
modulatory site, they have no activity when GABA is not simultaneously
binding to its agonist sites (compare Figare 9-23A and C).
So, how do benzodiazepines act as PAMs? This can occur only when GABA
is binding to its agonist sites. The combination of benzodiazepines at the
allosteric site plus GABA at its agonist sites increases the frequency of opening
of the chloride channel to an extent not possible with GABA alone (compare
Figure 9-23B and D).
The actions of benzodiazepines essentially as agonists at their positive
allosteric sites can be reversed by the neutral antagonist flumazenil (Figure 9-
24). Flumazenil is a short-acting intravenously adminis tered antagonist to
benzodiazepines that can reverse overdoses or anesthesia from benzodiazepines
but can also induce seizures or withdrawal in patients dependent upon

benzodiazepines.
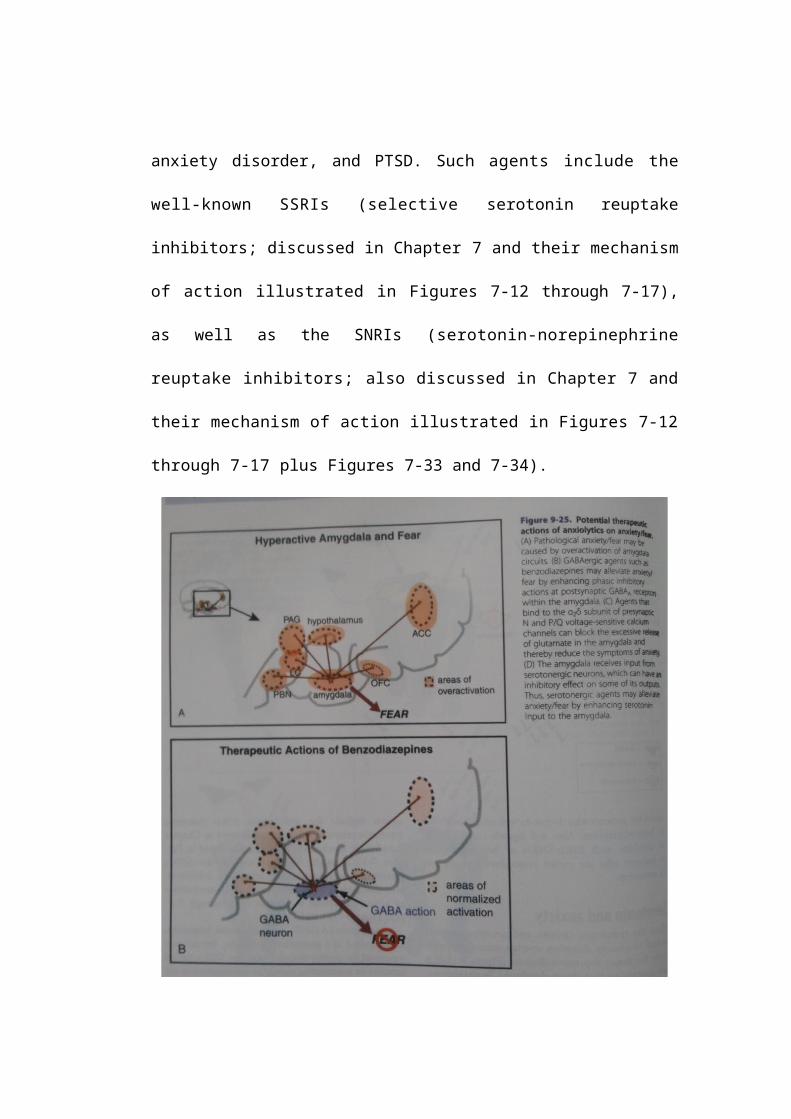
Benzodiazepines as anxiolytics
A simplified notion of how benzodiazepine anxiolytics might modulate
excessive output from the amygdala during fear responses in anxiety disorders
is shown in Figure 9-25. Excessive amygdala activity (shown in Figures 9-8
through 9-12 and in Figure 9-25A) is theoretically reduced by enhancing the
phasic inhibitory actions of benzodiazepine PAMs at postsynaptic GABAA
receptors within the amygdala to blunt fear-associated outputs, hypothetically
reducing the symptom of fear (Figure 9-25B). Benzodiazepines also theoretically
modulate excessive output from worry loops (Figure 9-26A) by enhancing the
actions of inhibitory interneurons in CSTC circuits (Figure 9-26B), hypothetically
reducing the symptom of worry.
Alpha-2-delta figands as anxiolytics
We have discussed voltage-sensitive calcium channel (VSCCs) in Chapter
3 and have illustrated presynaptic N and P/Q subtypes of VSCC and their
role in excitatory neurotransmitter release (see Figures 3-19 and 3-22
through 3-24). Gabapentin and pregabalin, also known as α2δ ligands, since
they bind to the α2δ subunit of presynaptic N and P/Q VSCCs, block the
release of excitatory neurotransmitters such as glutamate when
neurotransmission is excessive, as postulated in the amygdala to cause fear
(Figure 9-25A) and in CSTC circuits to cause worry (Figure 9-26A).

Hypothetically, α2δ ligands bind to open, overly active VSCCs in the
amygdala (Figure 9-25C) to reduce fear, and in CSTC circuits (Figure 9-26C) to
reduce worry. The α2δ ligands pregabalin and gabapentin have demonstrated
anxiolytic actions in social anxiety disorder and panic disorder, and are
also proven to be effective for the treatment of epilepsy and certain pain
conditions, including neuropathic pain and fibromyalgia. The actions of α2δ
ligands on VSCCs are discussed in Chapter 10 on pain and illustrated in
Figures 10-17 through 10-19. α2δ ligands clearly have different
mechanisms of action compared to serotonin reuptake inhibitors or
benzodiazepines, and thus can be useful for patients who do not do well on
SSRIs/SNRIs or benzodiazepines. Also, α2δ ligands can be useful to combine with
SSRIs/SNRIs or benzodiazepines in patients who are partial responders and
are not in remission.


Serotonin and anxiety
Since the symptoms, circuits, and neurotransmitters Liked to anxiety disorders
overlap extensively with those for major depressive disorder (Figure 9-1), it is
not surprising that drugs developed as antidepressants have proven to be
effective treatments for anxiety disorders. Indeed, the leading treatments for
anxiety disorders today are increasingly drugs originally developed as
antidepressants. Serotonin is a key neurotransmitter that innervates the
amygdala as well as all the elements of CSTC circuits, lamely, prefrontal cortex,
striatum, and thalamus, and thus is poised to regulate both fear and worry
serotonin pathways are discussed in Chapters 5 and 6 and illustrated in
Figure 6-33). Antidepressants that can increase serotonin output by locking
the serotonin transporter (SERT) are also effective in reducing symptoms of
anxiety and fear n every one of the anxiety disorders illustrated in Figures 9-2
though 9-5 - namely, GAD, panic disorder, social anxiety disorder, and PTSD.
Such agents include the well-known SSRIs (selective serotonin reuptake
inhibitors; discussed in Chapter 7 and their mechanism of action illustrated in
Figures 7-12 through 7-17), as well as the SNRIs (serotonin-norepinephrine
reuptake inhibitors; also discussed in Chapter 7 and their mechanism of
action illustrated in Figures 7-12 through 7-17 plus Figures 7-33 and 7-34).

A serotonin IA (5HT IA) partial agonist, buspirone, is recognized as a
generalized anxiolytic, but not as a treatment for anxiety disorder subtypes.
5HT1A partial agonists as augmenting agents to antidepressants are discussed in
Chapter 7, as are antidepressants combining 5HTIA partial agonism with serotonin
reuptake inhibition (i.e., SPARIs and vilazodone: see Figures 7-25 through 7-29),
which should theoretically be anxiolytic as well as antidepressant in action. The
5HTIA partial agonist properties of numerous atypical antipsychotics are discussed
in Chapter 5 and illustrated in Figures 5-15, 5-16, 5-25, and 5-26.
The potential anxiolytic actions of buspirone could theoretically be due to

5HTIA partial agonist actions at both presynaptic and postsynaptic 5HTIA receptors
(Figure 9-27 and Figures 5-15, 5-16, 5-25, 7-25 through 7-29), with actions at
both sites resulting in enhanced serotonergic activity in projections to the
amygdala (Figure 9-25D), prefrontal cortex, striatum, and thalamus (Figure 9-
26D). SSRIS and SNRIs theoretically do the same thing (Figures 9-25D and
9-26D). Since the onset of anxiolytic action for buspirone is delayed,
just as it is for antidepressants, this has led to the belief that 5HT 15
agonists exert their therapeutic effects by virtue of adaptive neuronal
events and receptor events (Figures 7-12 through 7-17 and 7-25 through 7-
29), rather than simply by the acute occupancy of 5HT1A receptors. In this
way, the presumed mechanism of action of 5HT IA partial agonists is analo-
gous to the antidepressants - which are also presumed to act by
adaptations in neurotransmitter receptors - and different from the
benzodiazepine anxiolytics - which act relatively acutely by occupying
benzodiazepine receptors.
Noradrenergic hyperactivity in anxiety
Norepinephrine is another neurotransmitter with important regulatory
input to the amygdala (Figure 9-28) and to the prefrontal cortex and
thalamus in CSTC circuits (Figure 9-29). Excessive noradrenergic output from
the locus coeruleus can not only result in numerous peripheral
manifestations of autonomic overdrive, as discussed above and illustrated
in Figures 9-8 through 9-12 but can also trigger numerous central symptoms

of anxiety and fear, such as nightmares. hvperarousal states, flashbacks,
and panic attacks (Figure 9-28A). Excessive noradrenergic activity can also
reduce the efficiency of information processing in the pre frontal cortex and
thus in CSTC circuits and theorettically cause worry (Figure 9-29A).
Hypothetically, these symptoms may be mediated in part by excessive
noradrenergic input onto α1 and β1 adrenergic Postsynaptic receptors in the
amygdala (Figure 9-28A) or prefrontal cortex (Figure 9-29A). Symptoms of
hyperarousal such as nightmares can be reduced in some patients with α1
adrenergic blockers such as Prazocin (Figure 9-28B); symptoms of fear
(Figure 9,28C) and worry (Figure 9-2913) can be reduced by norepinephrine
reuptake inhibitors (also called NET or norepinephrine transporter
inhibitors). The clinical effects of NET inhibitors can be confusing,
because symptoms of anxiety can be made transiently worse immediately
following initiation of an SNRI or selective NET inhibitor, when
noradrenergic activity is initially increased but the post-synaptic receptors
have not yet adapted. However, these same NET inhibitory actions, if
sustained over time, will down regulate and desensitize postsynaptic NE
receptors such as β1, receptors, and actually, reduce symptoms of fear and
worry long term (Figure 9-29B).

Fear conditioning versus fear extinction
Fear conditioning
Fear conditioning is a concept as old as Pavlov's dogs. If an aversive stimulus such
as foot shock is coupled with a neutral stimulus such as a bell, the animal learns to
associate the two and will develop fear when it hears a bell. In humans, fear is
learned during stressful experiences associated with emotional trauma and is
influenced by an individual’s genetic predisposition as well as by an individual’s
prior exposure to environmental stressors that can cause stress sensitization of
brain circuits (e.g., child abuse: see Chapter 6 and Figures 6-40 through 6-43).
Often, fearful situations are managed successfully and then forgotten. Some fears
are crucial for survival, such as appropriately fearing dangerous situations, and
thus the mechanism of learned fear, called fear conditioning, has been
extremely well conserved across species, including humans. However, other
fears that are “learned” and not “forgotten” may hypothetically progress to
anxiety disorders or a major depressive episode. This is a big problem, since
almost 30% of the population will develop an anxiety disorder, due in large
part to stressful environments, including exposure to fearful events during
normal activities, in twenty-first-century society, but in particular during
war and natural disasters. Hearing an explosion, smelling burning rubber,
seeing a picture of a Wounded civilian, and seeing or hearing flood waters are
all sensory experiences than can trigger traumatic re-experiencing and
generalized hyperarousal and fear in PTSD. Panic associated with social
situations Will “teach” the patient to panic in social situations in social
anxiety disorder. Panic randomly associated With an attack that happens to
occur in a crowd, on a bridge, or in a shopping center will also trigger
another panic attack when the same environment is encountered in panic

disorder. These and other symptoms of anxiety disorders are all forms of
learning known as fear conditioning (Figure 9-30).
The amygdala is involved in “remembering” the various stimuli
associated with a given fearful situation. It does this by increasing the
efficiency of neurotransmission at glutamatergic synapses in the lateral
amygdala as sensory input about those stimuli comes in from the thalamus
or sensory cortex (Figure 9-30). This input is then relayed to the central amygdala,
where fear conditioning also improves the efficiency of neurotransmission at
another glutamate synapse there (Figure 9-30). Both synapses are
restructured and permanent learning is embedded into this circuit by
NMDA receptors triggering long term potentiation and synaptic plasticity, so
that subsequent input from the sensory cortex and thalamus is very
efficiently processed to trigger the fear response as output from the
central amygdala every time there is sensory input associated with the ori -
ginal fearful event (Figure 9-30; see also Figures 9-8 through 9-13).

Input to the lateral amygdala is modulated by the prefrontal cortex, especially
the ventromedial prefrontal cortex (VMPFC), and by the hippocampus. If the
VMPFC is unable to suppress the fear response at the level of the amygdala,
fear conditioning proceeds. The hippocampus remembers the context of the fear
conditioning and makes sure fear is triggered when the fearful stimulus and all
its associated stimuli are, encountered. Most contemporary
psychopharmacological treatments for anxiety and fear act by suppressing the fear
output from the amygdale (figures 9-25 and 9-28) and therefore are not cures, sice
the fundamental neuronal learning underlying fear conditioning in these patient
remains in place.

