anu akt apoptosis
TRANSCRIPT

7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 1/14
doi:10.1152/ajpheart.00979.2007
294:H724-H735, 2008. First published 30 November 2007; Am J Physiol Heart Circ PhysiolMeetha MedhoraZhang, Garrett J. Gross, John R. Falck, Paresh K. Patel, Elizabeth R. Jacobs andAnuradha Dhanasekaran, Stephanie K. Gruenloh, J. Noelle Buonaccorsi, Rongprotect cardiomyocytes from hypoxia/anoxiapathway are activated by epoxyeicosatrienoic acids toMultiple antiapoptotic targets of the PI3K/Akt survival
You might find this additional info useful...
69 articles, 38 of which can be accessed free at:This article cites
http://ajpheart.physiology.org/content/294/2/H724.full.html#ref-list-1
10 other HighWire hosted articles, the first 5 are:This article has been cited by
[PDF][Full Text][Abstract], UNKNOWN, 2009; 297 (1): H37-H46. Am J Physiol Heart Circ Physiol
Myers, Douglas C. Rouse, Julie Foley, Abraham Nyska, Darryl C. Zeldin and John M. SeubertShayeganpour, Taibeh Orujy Jukar, J. Alyce Bradbury, Joan P. Graves, Laura M. DeGraff, PageYunfang Zhang, Haitham El-Sikhry, Ketul R. Chaudhary, Sri Nagarjun Batchu, AnooshirvanOverexpression of CYP2J2 provides protection against doxorubicin-induced cardiotoxicity
[PDF][Full Text][Abstract], July, 15 2009; 83 (2): 362-370.Cardiovasc Res
Joan P. Graves, Darryl C. Zeldin and John M. SeubertKetul R. Chaudhary, Sri Nagarjun Batchu, Dipankar Das, Mavanur R. Suresh, John R. Falck,post-ischaemic recovery of heart contractile functionRole of B-type natriuretic peptide in epoxyeicosatrienoic acid-mediated improved [PDF][Full Text][Abstract]
, June, 2010; 298 (6): H2201-H2207. Am J Physiol Heart Circ PhysiolNithipatikomGarrett J. Gross, John E. Baker, Anna Hsu, Hsiang-en Wu, John R. Falck and KasemheartsEvidence for a role of opioids in epoxyeicosatrienoic acid-induced cardioprotection in rat
[PDF][Full Text][Abstract], October, 2010; 299 (4): G833-G843. Am J Physiol Gastrointest Liver Physiol
Gregg L. Semenza and Da-Zhong XuShirhan Attan, Chirag D. Badami, Danielle Doucet, Dimitrios Barlos, Marta Bosch-Marce,Kolenkode B. Kannan, Sharvil U. Sheth, Francis J. Caputo, Qi Lu, Madhuri Ramanathan,Rena Feinman, Edwin A. Deitch, Anthony C. Watkins, Billy Abungu, Iriana Colorado,injuryHIF-1 mediates pathogenic inflammatory responses to intestinal ischemia-reperfusion
[PDF][Full Text], December, 15 2010; 588 (24): 4855-4856. J Physiol
Patrice Naud, Eduard Guasch and Stanley Nattelrelevance to clinical management
pathological cardiac electrical remodelling: potential basis andversusPhysiological
including high resolution figures, can be found at:Updated information and services
http://ajpheart.physiology.org/content/294/2/H724.full.html
can be found at: AJP - Heart and Circulatory PhysiologyaboutAdditional material and information
http://www.the-aps.org/publications/ajpheart
ISSN: 0363-6135, ESSN: 1522-1539. Visit our website at http://www.the-aps.org/.Physiological Society, 9650 Rockville Pike, Bethesda MD 20814-3991. Copyright © 2008 by the American Physiological Society.
intact animal to the cellular, subcellular, and molecular levels. It is published 12 times a year (monthly) by the Americanlymphatics, including experimental and theoretical studies of cardiovascular function at all levels of organization ranging from the
publishes original investigations on the physiology of the heart, blood vessels, and AJP - Heart and Circulatory Physiology

7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 2/14
This infomation is current as of March 17, 2011.
ISSN: 0363-6135, ESSN: 1522-1539. Visit our website at http://www.the-aps.org/.Physiological Society, 9650 Rockville Pike, Bethesda MD 20814-3991. Copyright © 2008 by the American Physiological Society.
intact animal to the cellular, subcellular, and molecular levels. It is published 12 times a year (monthly) by the Americanlymphatics, including experimental and theoretical studies of cardiovascular function at all levels of organization ranging from the
publishes original investigations on the physiology of the heart, blood vessels, and AJP - Heart and Circulatory Physiology

7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 3/14
Multiple antiapoptotic targets of the PI3K/Akt survival pathway are activated
by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia
Anuradha Dhanasekaran,1 Stephanie K. Gruenloh,1 J. Noelle Buonaccorsi,2 Rong Zhang,1
Garrett J. Gross,2 John R. Falck,4 Paresh K. Patel,4 Elizabeth R. Jacobs,1 and Meetha Medhora1
1
Division of Pulmonary and Critical Care Medicine, Cardiovascular Research Center, and 2
Department of Pharmacologyand Toxicology, 3 Medical College of Wisconsin, Milwaukee, Wisconsin; and 4 Department of Biochemistry, University of
Texas Southwestern Medical Center, Dallas, Texas
Submitted 23 August 2007; accepted in final form 28 November 2007
Dhanasekaran A, Gruenloh SK, Buonaccorsi JN, Zhang R, GrossGJ, Falck JR, Patel PK, Jacobs ER, Medhora M. Multiple antiapop-totic targets of the PI3K/Akt survival pathway are activated by epoxyei-cosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am J
Physiol Heart Circ Physiol 294: H724–H735, 2008. First publishedNovember 30, 2007; doi:10.1152/ajpheart.00979.2007.—Epoxyeicosa-trienoic acids (EETs) reduce infarction of the myocardium afterischemia-reperfusion injury to rodent and dog hearts mainly byopening sarcolemmal and mitochondrial potassium channels. Othermediators for the action of EET have been proposed, although nodefinitive pathway or mechanism has yet been reported. Using cul-tured cells from two rodent species, immortalized myocytes from amouse atrial lineage (HL-1) and primary myocytes derived fromneonatal rat hearts, we observed that pretreatment with EETs (1 Mof 14,15-, 11,12-, or 8,9-EET) attenuated apoptosis after exposure tohypoxia and reoxygenation (H/R). EETs also preserved the functionalbeating of neonatal myocytes in culture after exposure to H/R. Wedemonstrated that EETs increased the activity of the prosurvivalenzyme phosphatidylinositol 3-kinase (PI3K). In fact, cardiomyocytespretreated with EET and exposed to H/R exhibited antiapoptoticchanges in at least five downstream effectors of PI3K, protein kinaseB (Akt), Bcl-xL /Bcl-2-associated death promoter, caspases-9 and -3activities, and the expression of the X-linked inhibitor of apoptosis,
compared with vehicle-treated controls. The PI3K/Akt pathway is oneof the strongest intracellular prosurvival signaling systems. Our stud-ies show that EETs regulate multiple molecular effectors of thispathway. Understanding the targets of action of EET-mediated pro-tection will promote the development of these fatty acids as therapeu-tic agents against cardiac ischemia-reperfusion.
caspase; ischemia; rat; mouse; fatty acids
THE CYTOCHROME P-450-catalyzed epoxidation of arachidonicacid produces a series of epoxy fatty acids [epoxyeicosatrie-noic acids (EETs)] that influence a variety of cell and organfunctions. Cytochrome P-450-derived eicosanoids are pro-duced in a cell- and tissue-specific manner and have many
biological functions. EETs have been suggested to play diverseroles in cardiovascular homeostasis (25, 44, 48), the regulationof vascular tone, blood pressure, and ion transport and assecond messengers in numerous signaling pathways (5, 8, 19,52). More recently, these eicosanoids have been implicated inother critical biological processes including the control of cellproliferation, inflammation and homeostasis, migration, andplatelet aggregation (20, 45, 58, 64). EETs have been shown toplay an important role in the cardioprotection in rodents and
dogs subjected to ischemia-reperfusion (I/R) injury (55). Noclear mechanism for the action of EETs was derived in thesestudies. However, indirect evidence shows that the pharmaco-logical inhibition of mainly ATP- and Ca2-sensitive potas-sium channels (KATP, mitochondrial KATP, and KCa) (27, 43,54), and in some studies the p42/p44 mitogen-activated proteinkinase (55), phosphatidylinositol 3-kinase (PI3K) (54), andreactive oxygen species (27), attenuates the protection. Phar-
macological inhibition often nonspecifically affects signalingmolecules unrelated to the focus of investigation. Furthermore,some models were derived from mice in which the Ephx2 locuscoding for soluble epoxide hydrolase (sEH) was disrupted (54).Although knockout of this locus increases the levels of EETsby eliminating hydrolysis to dihydroxy derivatives, there aremultiple endogenous substrates for sEH, so that the exactintermediates which induce protection in these tissues may notbe limited to EETs. Currently, the opening of sarcolemmal andmitochondrial KATP channels is the most specific and favoredmechanism by which these fatty acids are believed to mediatemyocardial protection (27, 43, 54, 55).
The regulation of cell survival is crucial to the normalphysiology of multicellular organisms. The role of apoptosis inheart disease has been debated, although recent evidence sug-gests that it contributes to tissue injury in several cardiacdisorders including myocardial infarction, atherosclerosis,transplant, myocarditis, and cardiac failure. From 5% to 30%of cardiac myocytes undergo apoptosis in rodent and humanswithin 16 h of reperfusion (2, 21, 47, 50, 60), and this trendpersists for months (up to 0.25% in diseased humans comparedwith up to 0.002% in controls) (28, 46, 60). In intact animals,apoptosis occurs during myocardial infarction, heart failure,and transplant injuries (13). Genetic approaches using rodentshave determined that inhibition of apoptosis in cardiomyocytesreduces infarct size by 50–70% (9, 37, 50). Apoptotic rates aslow as 0.023% are sufficient to cause lethal dilated cardiomy-
opathy in transgenic mice that are engineered to have cardiac-restricted expression of caspase-8. This rate of apoptosis is 5-to 10-fold lower than that measured in cardiac tissue frompatients with end-stage heart failure (13, 46). A recent findingin a renal proximal tubular epithelial cell line demonstrated thatEETs inhibit apoptosis induced by serum withdrawal andetoposide (6, 41). We have reported that EETs inhibit apoptosisinduced by the engagement of the death receptor Fas or byserum withdrawal in human vascular endothelial cells cultured
Address for reprint requests and other correspondence: M. Medhora, Car-diovascular Center, MEB M4870, Medical College of Wisconsin, 8701Watertown Plank Rd., Milwaukee, WI 53226 (e-mail: [email protected]).
The costs of publication of this article were defrayed in part by the paymentof page charges. The article must therefore be hereby marked “advertisement ”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Am J Physiol Heart Circ Physiol 294: H724–H735, 2008.First published November 30, 2007; doi:10.1152/ajpheart.00979.2007.
0363-6135/08 $8.00 Copyright © 2008 the American Physiological Society http://www.ajpheart.orgH724
7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 4/14
from the heart or lung (17, 36). Hence, we were interested tosee whether EETs have the same antiapoptotic effect in cardio-myocytes after I/R since myocardial cell death caused byapoptosis is a main feature of this condition.
We investigated four different end points to report whetherEETs inhibit cell death/apoptosis in cultured cardiomyocytes,which are subjected to hypoxia and reoxygenation (H/R) to
simulate conditions of I/R. We describe for the first time theactivation of the enzyme PI3K by EETs in cultured myocytes.We also systematically followed multiple downstream anti-apoptotic signals, including phosphorylation of protein kinaseB (Akt) and bcl-xl/bcl-2-associated death promoter (BAD),increased intracellular levels of X-linked inhibitor of apoptosis(XIAP), and decreased activity of caspases-9 and -3. Ourresults support at least five prosurvival effectors that maymediate the EET-induced protection of cardiomyocytes viastimulation of the PI3K/Akt pathway.
MATERIALS AND METHODS
Culture of Neonatal Rat Ventricular Myocytes
Animals were housed in the accredited Biomedical Resource Cen-ter. All procedures for animals in this study were reviewed andapproved by the Institutional Animal Care and Use Committee. Heartsfrom 1-day-old Sprague-Dawley neonatal rats were excised, and theventricular myocardium was cut into small pieces (2 mm3) indissociation buffer containing (in mm) 116 NaCl, 20 HEPES, 1NaH2PO4, 5.5 glucose, 5.4 KCl, and 0.8 MgSO4 and 0.6 ml/l phenolred (pH 7.35) with 0.15 mg/ml collagenase (Worthington II, Lake-wood, NJ) and 0.52 mg/ml pancreatin (Life Technologies, GrandIsland, NY) and incubated on a shaker at 37°C for 20 min at 100 rpm.Tissue pieces were allowed to settle, and the supernatant (containingmyocytes) was collected, suspended in 1 ml newborn calf serum(GIBCO, Carlsbad, CA), and centrifuged at 1,000 rpm for 6 min. Thecell pellet was resuspended in 1 ml newborn calf serum and stored at37°C. The procedure was repeated until all tissue was digested. The
cells were then resuspended in Dulbecco’s modified Eagle’s medium(DMEM) supplemented with 17% medium 199, 10% horse serum, 5%fetal bovine serum (FBS), 0.5% penicillin-streptomycin, and 20 mmHEPES (pH 7.2) for 2 h. This facilitates the separation of ventricularmyocytes from the faster-attaching nonmyocytes. The ventricularmyocytes in the supernatant were collected and plated on gelatin-coated dishes. They were cultured in media containing bromode-oxyuridine (5-bromo-2-deoxyuridine) at a final concentration of 0.1mM. The cells were used for experiments after demonstrating rhyth-mic contractions (48–72 h).
HL-1 Cells
Cells of HL-1, a cardiac muscle cell line derived from the AT-1mouse atrial myocyte tumor lineage, were a gift from Dr. William C.
Claycomb (Louisiana State University Health Sciences Center, NewOrleans, LA) and maintained accordingly as described (11). HL-1 cellswere used for experimentation after reaching 70–80% confluence.
Treatment of Cells
Rat neonatal myocytes were cultured to 70% confluency and thenserum starved in basal medium (DMEM 0.1% BSA) for 24 h for theexperiments in Fig. 1. The cells were stimulated with one of fourconcentrations of 14,15-EET (0.1, 0.3, 1, or 10 M) or vehicle(ethanol) and lysed as described in Western Blot Analysis. For otherexperiments, separate groups of neonatal rat ventricular myocytes ormouse atrial HL-1 cells received no intervention (normoxia controls)or were exposed to H/R after pretreatment with two applications of either vehicle (ethanol) or a single regioisomer of EET (1 M of
14,15-, 11,12-, or 8,9-EET). Exposure to EET was accomplished 16 h
before (first application) and just before (second application) H/R.Hypoxia was simulated for 8 h in neonatal myocytes by exposure to5% CO2-95% N2 in an airtight chamber in the presence of serum- andglucose-free DMEM containing 10 mM deoxyglucose to inhibitglycolysis. The levels of oxygen were monitored continuously andwere below 2% at all times. Reoxygenation was performed for 16 h inDMEM 10% FBS except if otherwise mentioned [for PI3K activityor analyses of phospho-Akt (pAkt)]. HL-1 cells were treated similarlyexcept that deoxyglucose was not included in the serum-free medium.In some experiments, the PI3K inhibitor wortmannin (WT) was added30 min before each application of EET. Control cells that received nointervention or H/R were maintained in DMEM 10% FBS through-out the duration of the experiments.
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide
(MTT) Assay
The cells (HL-1 and neonatal myocytes) were cultured in 60-mmdishes (plated at a density approximately half million cells) to 80%confluency and then maintained in either complete medium orswitched to basal medium after 42 h. The samples were maintainedunder normoxia or treated with a single regioisomer of EET or vehicleand subjected to H/R as described in Treatment of Cells. At the end of the incubation period, the survival of the cells was determined by the3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)assay as described in the manufacturer’s protocol (Cat. No. V-13154;Molecular Probes, Eugene, OR). Briefly, the cells were incubated for3 h in phenol red-free medium containing 0.5% of the yellowmitochondrial dye MTT. We used a slightly higher confluency(80%) in the MTT protocol since the assay required cell viability for
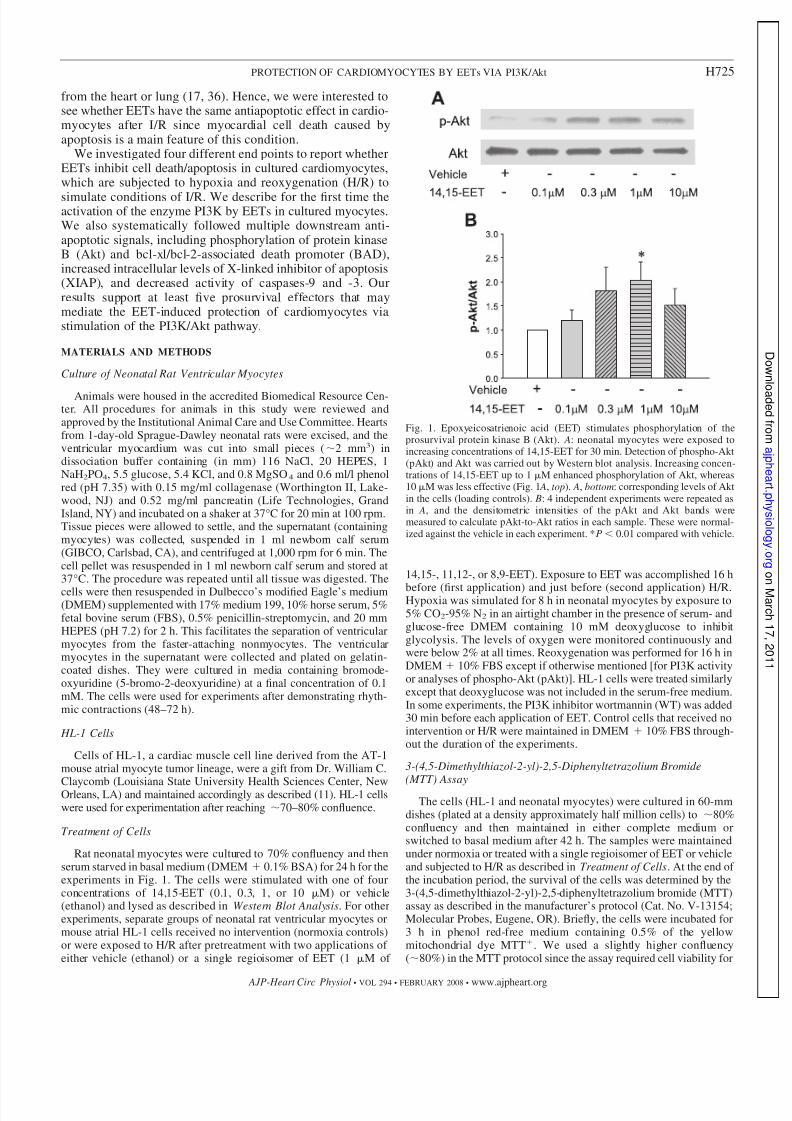
Fig. 1. Epoxyeicosatrienoic acid (EET) stimulates phosphorylation of theprosurvival protein kinase B (Akt). A: neonatal myocytes were exposed toincreasing concentrations of 14,15-EET for 30 min. Detection of phospho-Akt(pAkt) and Akt was carried out by Western blot analysis. Increasing concen-trations of 14,15-EET up to 1 M enhanced phosphorylation of Akt, whereas10 M was less effective (Fig. 1 A, top). A, bottom: corresponding levels of Aktin the cells (loading controls). B: 4 independent experiments were repeated asin A, and the densitometric intensities of the pAkt and Akt bands weremeasured to calculate pAkt-to-Akt ratios in each sample. These were normal-ized against the vehicle in each experiment. *P 0.01 compared with vehicle.
H725PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org

7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 5/14
an additional 3 h of incubation with the substrate after exposure toH/R. The amount of blue formazan dye generated from MTT wasproportional to the number of live cells. The MTT reaction wasterminated by the addition of DMSO to the medium, followed byincubation for 10 min at 37°C. The absorbance was read at 540 nm ina spectrophotometer. The values of the reaction were obtained afterthe subtraction of matched blanks, and the optical densities of thecontrols were taken as 100% for comparisons with values for other
samples. The readings for the test cells were expressed as percentagesof control.
Hoechst Staining
The cells were cultured in six-well plates to 70% confluency,treated with EETs, and subjected to H/R as described for the MTTassay in Treatment of Cells. They were then stained with 1 l of Hoechst 33342 (5 mg/ml; Cat. No. V-13244, Molecular Probes) in 1ml basal medium and incubated for 30 min. Stained cells were washedtwice with PBS (Sigma, St. Louis, MO) and imaged under a fluores-cent microscope (excitation, 350 nm; emission, 460 nm).
Annexin V Binding
Annexin V binds to phosphatidylserine, which appears in the outerleaflet of the plasma membrane in early apoptotic cells. Cells (neo-natal myocytes and HL-1 cells) were cultured in 60-mm dishes to 70%confluency and subjected to normoxia or H/R with or without pre-treatment with EET as described in Treatment of Cells. The cells werewashed with PBS and treated with FITC-labeled annexin V (0.2g/ml) for 20 min at room temperature (17). Labeling with FITC-coupled annexin V was performed according to the manufacturer’sprotocol (BD Biosciences, San Diego, CA) as previously described(17). The labeled cells (10,000/sample) were analyzed by measuringfluorescence intensity using a FACScan flow cytometer (BectonDickinson) in conjunction with CellQuest software (BD Biosciences).
Activity of Caspases-3 and -9
Cells (neonatal myocytes and HL-1 cells) were cultured in 60-mmdishes to 70% confluency and subjected to H/R or normoxia with orwithout pretreatment with EET as described in Treatment of Cells.The cells were collected by centrifugation, washed twice with ice-coldPBS, resuspended in lysis buffer containing (in mM) 5 MgCl2, 1EGTA, 0.1% Triton X-100, and 25 HEPES (pH 7.5), and storedovernight at 80°C as previously described (28). The release of free7-amino-4-trifluoromethyl coumarin (AFC) from the synthetic sub-strate N -acetyl-Asp-Glu-Val-Asp-AFC (50 M) for caspase-3 and therelease of free AFC from the N -acetyl-Leu-Glu-His-Asp-AFC sub-strate for caspase-9 at 37°C were determined by fluorescence mea-surements in a SpectraFluor Plus plate reader after excitation atwavelength of 390 nm. Emission was measured at 535 nm.
Detection of Cleaved Caspase-3 Activity
Cells were treated under normoxia or H/R with or without EET,and cleaved caspase was detected according to the manufacturer’sprotocol (Cell Signaling, Charlottesville, VA). Briefly, cells weretreated as described in Treatment of Cells, and at the end of thetreatment time, cells were washed with PBS and fixed in PBScontaining 3% paraformaldehyde for 20 min at 4°C. The cells werethen washed three times with Tris-buffered saline-Tween 20 [TBST;10 mM Tris HCl (pH 8.0), 150 mM NaCl, and 0.05% Tween 20] for5 min each time and incubated with 1 ml blocking buffer for 45–60min. The cells were rinsed and incubated in cleaved caspase antibody(1:200 dilution) in TBST/BSA for 24 h at 4°C. They were washedthree times, incubated with appropriate fluorescent secondary anti-body, and detected using a FITC filter (excitation 490 nm; emission525 nm).
PI3K Activity
PI3K was estimated according to the manufacturer’s protocol(Echelon, Salt Lake City, UT). Briefly, cells were pretreated withEET, subjected to hypoxia for 8 h, and reoxygenated for 60 min. Thecells were then scraped in ice-cold lysis buffer containing (in mM) 137NaCl, 20 Tris HCl (pH 7.4), 1 CaCl2, and 1 MgCl2 and 0.1 mM sodiumorthovanadate with 1% Nonidet P-40 (NP-40) and 1 mM PMSF. The
lysate was centrifuged, and the supernatant was incubated with 5 lanti-PI3K antibody for 1 h. Protein A agarose beads (60 l of a 50%slurry) were added, and the lysates were mixed gently for 1 h at 4°C.The immunoprecipitated enzyme was collected by centrifugation for5 s at 3,000 g and washed once with the lysis buffer and again withbuffer of (in mM) 10 Tris HCl (pH 7.4), 150 NaCl, and 5 EDTAcontaining 0.1 mM sodium orthovanadate. The phosphatidylinositol4,5-biophosphate [PI(4,5)P2] substrate was added to the enzyme alongwith reaction buffer and left at room temperature for 3 h. The kinasereaction was stopped by the addition of 2.5 l 100 mM EDTA. Theplate was then set with standards, blanks, and samples, incubated withthe detector protein, and read using a plate reader at 450 nm.
Immunofluorescence Studies for pAkt
Neonatal rat ventricular myocytes were plated on collagen-coatedcoverslips, pretreated with EET, and exposed to H/R or normoxia for8 h as described in Detection of Cleaved Caspase-3 Activity. Afterreoxygenation for 60 min, the cells were fixed in 4% paraformalde-hyde, permeabilized in methanol (20°C), and incubated for 30 minat 37°C with primary monoclonal antibody for pAkt (Ser473; CellSignaling, Beverly, MA) at a dilution of 1:200 in PBS (0.5). Thesamples were washed and incubated with anti-mouse biotinylatedsecondary antibodies (1:200 dilution; Santa Cruz Biotechnology) for30 min at 37°C. After being washed with 0.5 PBS, the cells wereincubated for 15 min at 37°C with avidin-conjugated 1:500 FITC,followed by 1:1,000 4’,6-diamidino-2-phenylindole (DAPI) (to stainthe nuclei) for 5 min at room temperature. The samples were washedagain with 0.5 PBS and mounted on microscopic slides. Imageswere captured using confocal microscopy using appropriate filters forvisualization of FITC (described above) and DAPI (excitation, ultra-violet; emission, 461 nm).
Western Blot Analysis
HL-1 cells and neonatal myocytes were cultured in 35-mm dishesto 70% confluency, washed, and incubated with basal medium for24 h. The cells were treated under normoxia or H/R with or withoutEETs. They were kept on ice and washed three times with cold PBS.Proteins were solubilized and extracted with 50 l buffer of 50 mM Tris(pH 8.0), 150 mM NaCl, 0.5% SDS, 1% NP-40, 0.5% sodium deoxy-cholate, 1 mM EDTA, 1 protease inhibitor cocktail (Pharmingen, SanDiego, CA), and 1 phosphatase inhibitors (Calbiochem, San Diego,CA). The lysates were used to estimate protein content with theBio-Rad DC Protein Assay reagent (Bio-Rad, Hercules, CA). Equalamounts of protein (10–60 g) from each sample were electropho-
resed on a 10% SDS-PAGE with running buffer and transferred tonitrocellulose as described (17, 36). The membranes were treatedwith primary antibody for XIAP, pBAD, BAD, or pAkt (1:1,000dilution; Cell Signaling) for 18 h. They were again washed threetimes before incubating with matched secondary antibody (1:5,000) for 45 min. The protein bands were developed with en-hanced chemiluminescence reagents (ECL or ECL plus; GEHealthcare, Piscataway, NJ).
Functional Evaluation of Contractility of Neonatal Myocytes
Cultured neonatal myocytes (5 days old) were washed, incubatedwith basal medium for 8 h, treated with vehicle or EET, and furtherincubated overnight. The cells were treated under normoxia or ex-posed to hypoxia for specific times after pretreatment with two
H726 PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org
7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 6/14
applications of either vehicle or a regioisomer of EET (1 M of 14,15-or 11,12-EET). In pilot experiments, vehicle-treated cells stoppedbeating between 30 min and up to 8 h of exposure to anoxicconditions. Test plates from each batch were incubated under hypoxiaand observed at half-hour intervals. When 85% vehicle-treated cellsmaintained in anoxic conditions stopped beating, all test and controlgroups were exposed to 1 h of reoxygenation. The dishes wereremoved from the incubator and immediately observed under a light
microscope. Islands of beating cells were counted in a blinded mannerin at least four fixed fields/dish from four independent batches of cells.The frequency of beating of at least 25 cells/treatment was alsocounted in the same samples using a stopwatch. The average numberof beating cells/field and the average frequency (beats/min) for eachtreatment were recorded, decoded, and then used to calculate the meanand SE of the mean for statistical analysis.
Statistical Comparisons
All values are expressed as means SE from at least three or moresamples in each experiment. Comparisons between controls andtreatments were analyzed by ANOVA, followed by Tukey’s test whenpermitted. Values for P 0.05 were considered significant.
RESULTS
EETs Induce Phosphorylation of Akt in Neonatal, Rat,and Cardiac Myocytes
Primary cultures of myocytes were stimulated with in-creasing concentrations of 14,15-EET to test the effect of the fatty acid on activating the prosurvival kinase Akt (Fig.1). After 30 min of treatment, the cells were lysed andanalyzed by Western blot analysis to determine pAkt levels.As seen in Fig. 1 A, there is an increase in pAkt (top)compared with Akt (bottom) in EET-treated cells. Analysesfrom four independent experiments demonstrated a signifi-cant increase in the densitometric ratio of pAkt to Aktversus the vehicle-treated cells at the 1 M concentration of
14,15-EET (n 4; Fig. 1 B). At an even higher concentra-tion of 14,15-EET (10 M), the ratio was lower than for 1M of 14,15-EET, as shown in Fig. 1. In cells treated with10 M EET, this ratio was statistically indistinguishable tothat of the vehicle.
EETs Protect Mouse Atrial (HL-1) and Rat NeonatalCardiac Myocytes Against H/R-Induced Apoptosis
MTT cell viability assay. Both cell types (HL-1 and primaryneonatal cardiomyocytes) showed increased survival rates when
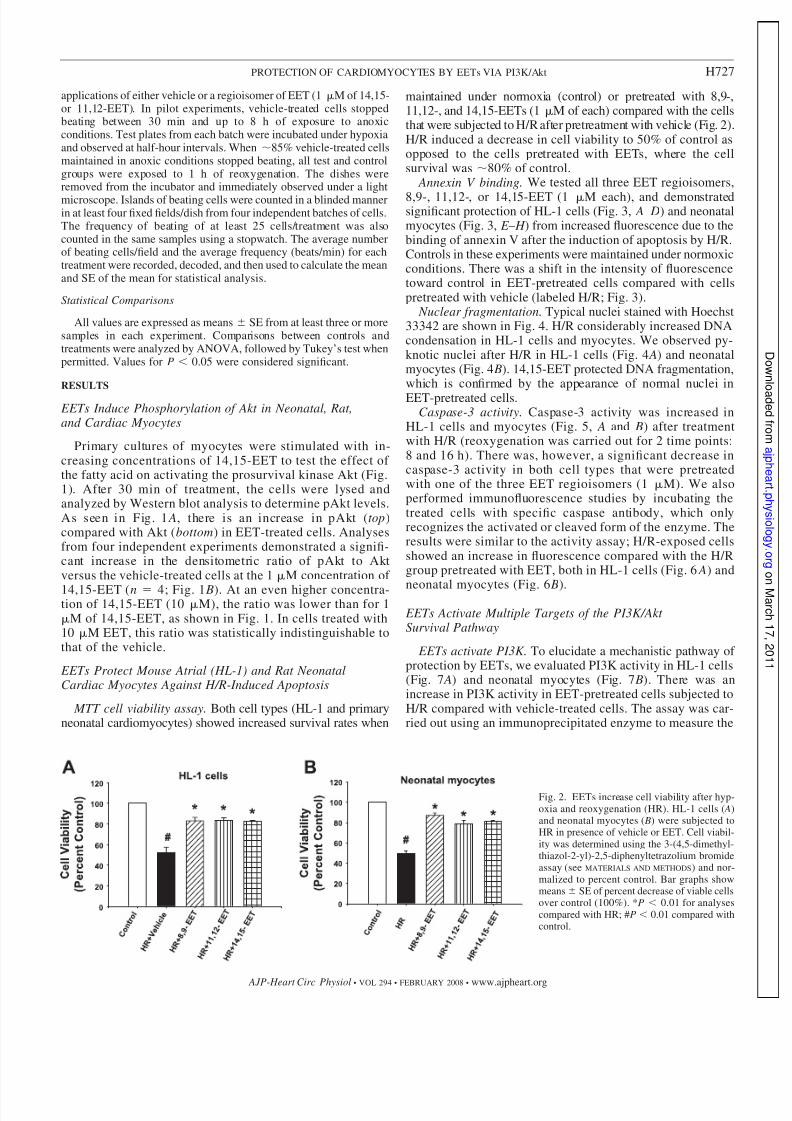
maintained under normoxia (control) or pretreated with 8,9-,11,12-, and 14,15-EETs (1 M of each) compared with the cellsthat were subjected to H/R after pretreatment with vehicle (Fig. 2).H/R induced a decrease in cell viability to 50% of control asopposed to the cells pretreated with EETs, where the cellsurvival was 80% of control.
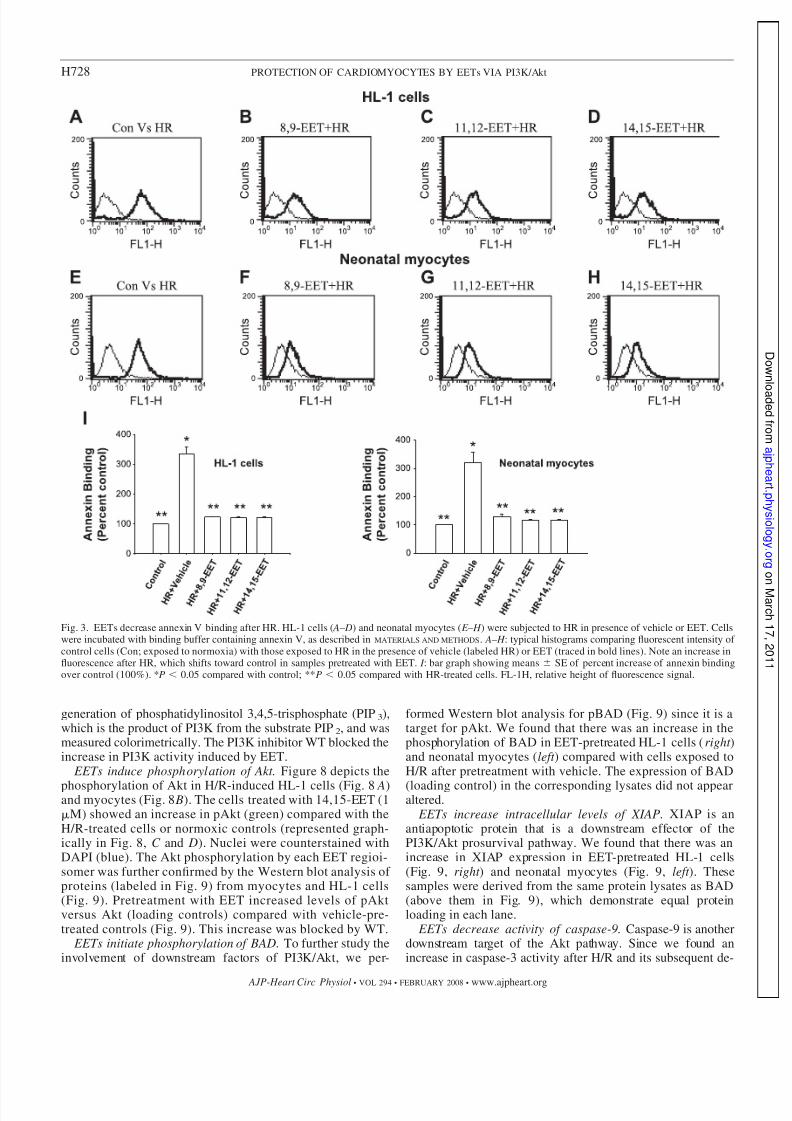
Annexin V binding. We tested all three EET regioisomers,
8,9-, 11,12-, or 14,15-EET (1 M each), and demonstratedsignificant protection of HL-1 cells (Fig. 3, A– D) and neonatalmyocytes (Fig. 3, E – H ) from increased fluorescence due to thebinding of annexin V after the induction of apoptosis by H/R.Controls in these experiments were maintained under normoxicconditions. There was a shift in the intensity of fluorescencetoward control in EET-pretreated cells compared with cellspretreated with vehicle (labeled H/R; Fig. 3).
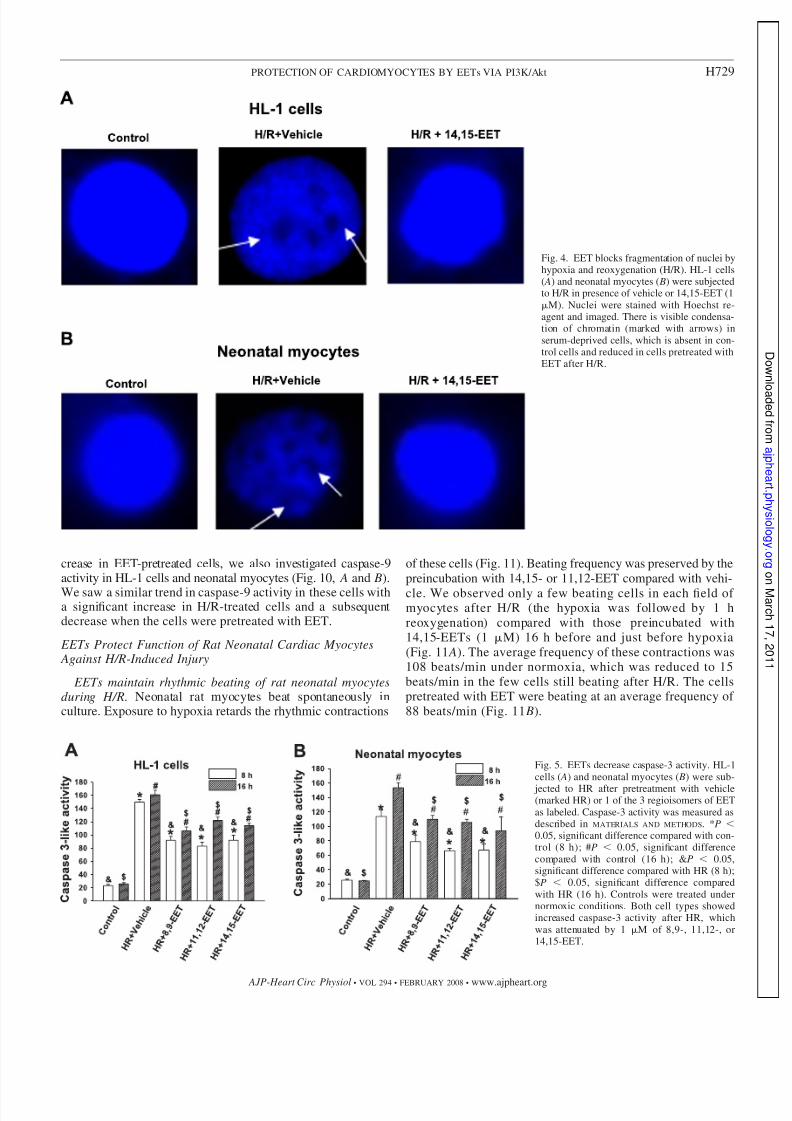
Nuclear fragmentation. Typical nuclei stained with Hoechst33342 are shown in Fig. 4. H/R considerably increased DNAcondensation in HL-1 cells and myocytes. We observed py-knotic nuclei after H/R in HL-1 cells (Fig. 4 A) and neonatalmyocytes (Fig. 4 B). 14,15-EET protected DNA fragmentation,which is confirmed by the appearance of normal nuclei inEET-pretreated cells.
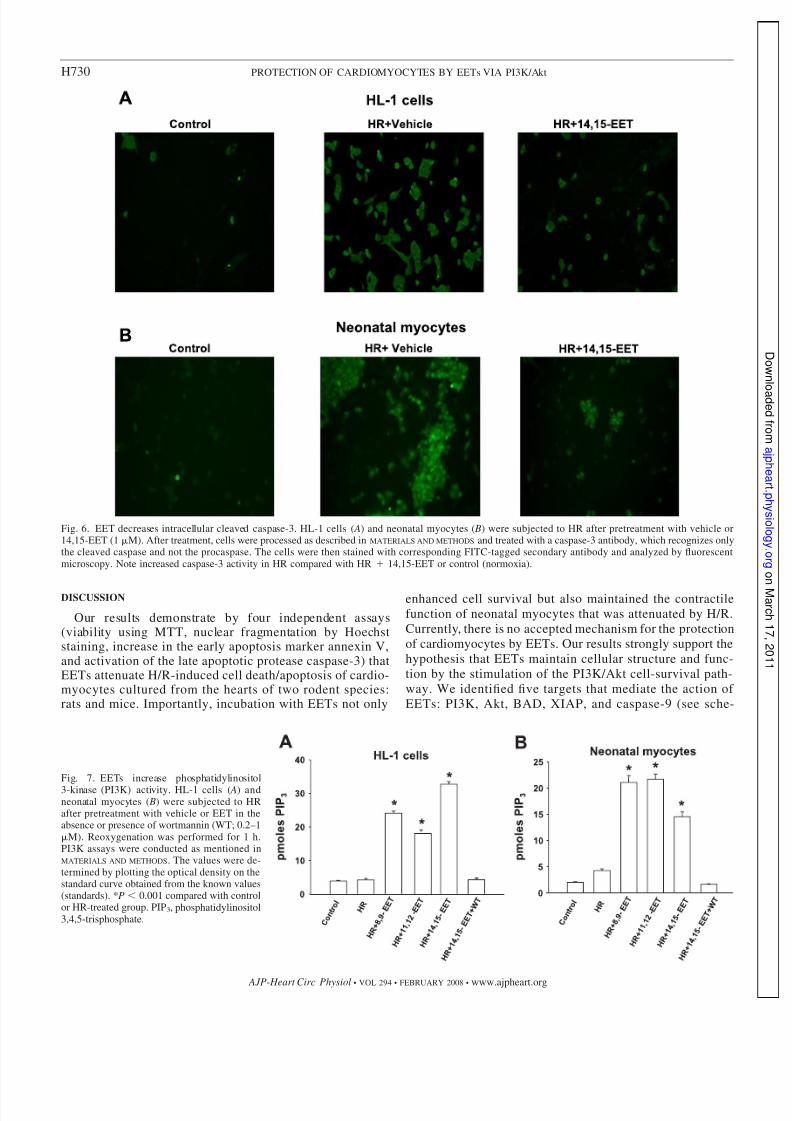
Caspase-3 activity. Caspase-3 activity was increased inHL-1 cells and myocytes (Fig. 5, A and B) after treatmentwith H/R (reoxygenation was carried out for 2 time points:8 and 16 h). There was, however, a significant decrease incaspase-3 activity in both cell types that were pretreatedwith one of the three EET regioisomers (1 M). We alsoperformed immunofluorescence studies by incubating thetreated cells with specific caspase antibody, which onlyrecognizes the activated or cleaved form of the enzyme. Theresults were similar to the activity assay; H/R-exposed cellsshowed an increase in fluorescence compared with the H/Rgroup pretreated with EET, both in HL-1 cells (Fig. 6 A) and
neonatal myocytes (Fig. 6 B).
EETs Activate Multiple Targets of the PI3K/Akt Survival Pathway
EETs activate PI3K. To elucidate a mechanistic pathway of protection by EETs, we evaluated PI3K activity in HL-1 cells(Fig. 7 A) and neonatal myocytes (Fig. 7 B). There was anincrease in PI3K activity in EET-pretreated cells subjected toH/R compared with vehicle-treated cells. The assay was car-ried out using an immunoprecipitated enzyme to measure the
Fig. 2. EETs increase cell viability after hyp-oxia and reoxygenation (HR). HL-1 cells ( A)and neonatal myocytes ( B) were subjected toHR in presence of vehicle or EET. Cell viabil-ity was determined using the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromideassay (see MATERIALS AND METHODS) and nor-malized to percent control. Bar graphs showmeans SE of percent decrease of viable cellsover control (100%). *P 0.01 for analysescompared with HR; #P 0.01 compared withcontrol.
H727PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org
7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 7/14
generation of phosphatidylinositol 3,4,5-trisphosphate (PIP3),which is the product of PI3K from the substrate PIP2, and wasmeasured colorimetrically. The PI3K inhibitor WT blocked theincrease in PI3K activity induced by EET.
EETs induce phosphorylation of Akt. Figure 8 depicts the
phosphorylation of Akt in H/R-induced HL-1 cells (Fig. 8 A)and myocytes (Fig. 8 B). The cells treated with 14,15-EET (1M) showed an increase in pAkt (green) compared with theH/R-treated cells or normoxic controls (represented graph-ically in Fig. 8, C and D). Nuclei were counterstained withDAPI (blue). The Akt phosphorylation by each EET regioi-somer was further confirmed by the Western blot analysis of proteins (labeled in Fig. 9) from myocytes and HL-1 cells(Fig. 9). Pretreatment with EET increased levels of pAktversus Akt (loading controls) compared with vehicle-pre-treated controls (Fig. 9). This increase was blocked by WT.
EETs initiate phosphorylation of BAD. To further study theinvolvement of downstream factors of PI3K/Akt, we per-
formed Western blot analysis for pBAD (Fig. 9) since it is atarget for pAkt. We found that there was an increase in thephosphorylation of BAD in EET-pretreated HL-1 cells (right )and neonatal myocytes (left ) compared with cells exposed toH/R after pretreatment with vehicle. The expression of BAD
(loading control) in the corresponding lysates did not appearaltered.
EETs increase intracellular levels of XIAP. XIAP is anantiapoptotic protein that is a downstream effector of thePI3K/Akt prosurvival pathway. We found that there was anincrease in XIAP expression in EET-pretreated HL-1 cells(Fig. 9, right ) and neonatal myocytes (Fig. 9, left ). Thesesamples were derived from the same protein lysates as BAD(above them in Fig. 9), which demonstrate equal proteinloading in each lane.
EETs decrease activity of caspase-9. Caspase-9 is anotherdownstream target of the Akt pathway. Since we found anincrease in caspase-3 activity after H/R and its subsequent de-
Fig. 3. EETs decrease annexin V binding after HR. HL-1 cells ( A– D) and neonatal myocytes ( E – H ) were subjected to HR in presence of vehicle or EET. Cellswere incubated with binding buffer containing annexin V, as described in MATERIALS AND METHODS. A– H : typical histograms comparing fluorescent intensity of control cells (Con; exposed to normoxia) with those exposed to HR in the presence of vehicle (labeled HR) or EET (traced in bold lines). Note an increase influorescence after HR, which shifts toward control in samples pretreated with EET. I : bar graph showing means SE of percent increase of annexin bindingover control (100%). *P 0.05 compared with control; **P 0.05 compared with HR-treated cells. FL-1H, relative height of fluorescence signal.
H728 PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org
7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 8/14
crease in EET-pretreated cells, we also investigated caspase-9activity in HL-1 cells and neonatal myocytes (Fig. 10, A and B).
We saw a similar trend in caspase-9 activity in these cells witha significant increase in H/R-treated cells and a subsequentdecrease when the cells were pretreated with EET.
EETs Protect Function of Rat Neonatal Cardiac Myocytes Against H/R-Induced Injury
EETs maintain rhythmic beating of rat neonatal myocytesduring H/R. Neonatal rat myocytes beat spontaneously inculture. Exposure to hypoxia retards the rhythmic contractions
of these cells (Fig. 11). Beating frequency was preserved by thepreincubation with 14,15- or 11,12-EET compared with vehi-
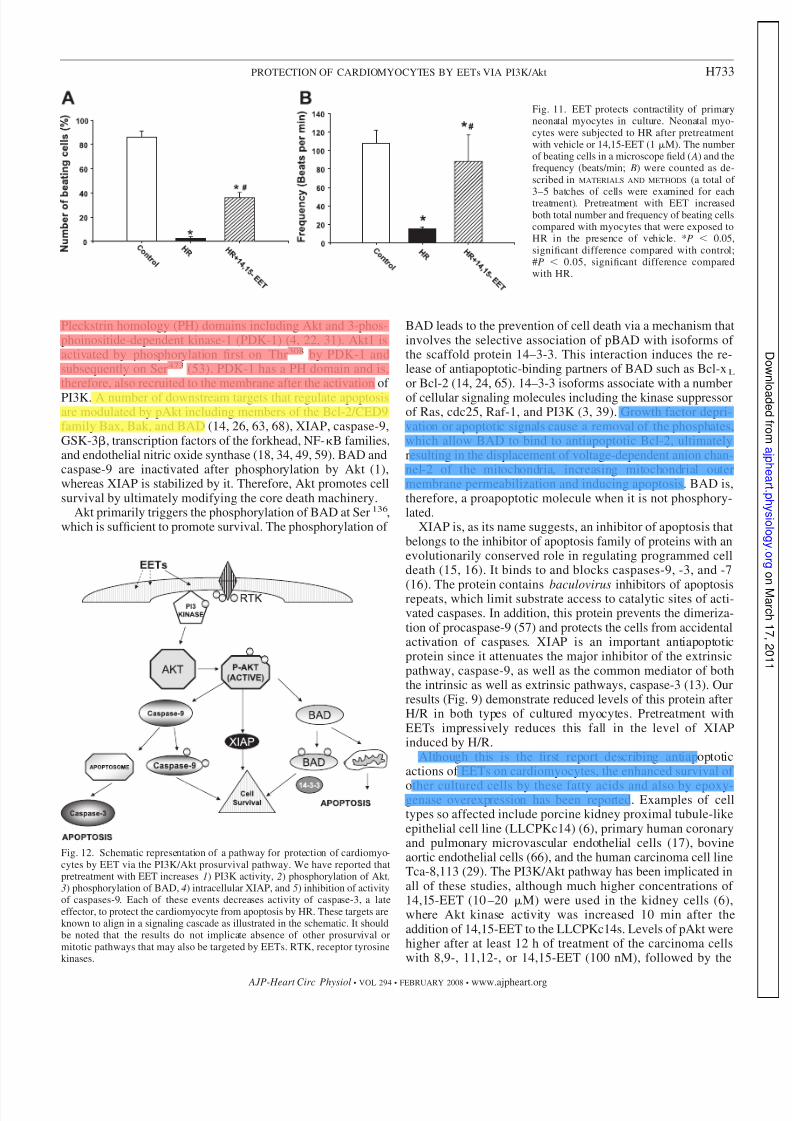
cle. We observed only a few beating cells in each field of myocytes after H/R (the hypoxia was followed by 1 hreoxygenation) compared with those preincubated with14,15-EETs (1 M) 16 h before and just before hypoxia(Fig. 11 A). The average frequency of these contractions was108 beats/min under normoxia, which was reduced to 15beats/min in the few cells still beating after H/R. The cellspretreated with EET were beating at an average frequency of 88 beats/min (Fig. 11 B).
Fig. 4. EET blocks fragmentation of nuclei byhypoxia and reoxygenation (H/R). HL-1 cells( A) and neonatal myocytes ( B) were subjectedto H/R in presence of vehicle or 14,15-EET (1M). Nuclei were stained with Hoechst re-agent and imaged. There is visible condensa-tion of chromatin (marked with arrows) inserum-deprived cells, which is absent in con-trol cells and reduced in cells pretreated withEET after H/R.
Fig. 5. EETs decrease caspase-3 activity. HL-1cells ( A) and neonatal myocytes ( B) were sub-
jected to HR after pretreatment with vehicle(marked HR) or 1 of the 3 regioisomers of EETas labeled. Caspase-3 activity was measured asdescribed in MATERIALS AND METHODS. *P 0.05, significant difference compared with con-trol (8 h); #P 0.05, significant differencecompared with control (16 h); &P 0.05,significant difference compared with HR (8 h);$P 0.05, significant difference comparedwith HR (16 h). Controls were treated undernormoxic conditions. Both cell types showedincreased caspase-3 activity after HR, whichwas attenuated by 1 M of 8,9-, 11,12-, or14,15-EET.
H729PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org
7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 9/14
DISCUSSION
Our results demonstrate by four independent assays(viability using MTT, nuclear fragmentation by Hoechststaining, increase in the early apoptosis marker annexin V,and activation of the late apoptotic protease caspase-3) thatEETs attenuate H/R-induced cell death/apoptosis of cardio-myocytes cultured from the hearts of two rodent species:rats and mice. Importantly, incubation with EETs not only
enhanced cell survival but also maintained the contractilefunction of neonatal myocytes that was attenuated by H/R.Currently, there is no accepted mechanism for the protectionof cardiomyocytes by EETs. Our results strongly support thehypothesis that EETs maintain cellular structure and func-tion by the stimulation of the PI3K/Akt cell-survival path-way. We identified five targets that mediate the action of EETs: PI3K, Akt, BAD, XIAP, and caspase-9 (see sche-
Fig. 6. EET decreases intracellular cleaved caspase-3. HL-1 cells ( A) and neonatal myocytes ( B) were subjected to HR after pretreatment with vehicle or14,15-EET (1 M). After treatment, cells were processed as described in MATERIALS AND METHODS and treated with a caspase-3 antibody, which recognizes onlythe cleaved caspase and not the procaspase. The cells were then stained with corresponding FITC-tagged secondary antibody and analyzed by fluorescentmicroscopy. Note increased caspase-3 activity in HR compared with HR 14,15-EET or control (normoxia).
Fig. 7. EETs increase phosphatidylinositol3-kinase (PI3K) activity. HL-1 cells ( A) andneonatal myocytes ( B) were subjected to HRafter pretreatment with vehicle or EET in theabsence or presence of wortmannin (WT; 0.2–1M). Reoxygenation was performed for 1 h.PI3K assays were conducted as mentioned inMATERIALS AND METHODS. The values were de-termined by plotting the optical density on thestandard curve obtained from the known values(standards). *P 0.001 compared with controlor HR-treated group. PIP3, phosphatidylinositol3,4,5-trisphosphate.
H730 PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org

7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 10/14
matic in Fig. 12). Each one of these proteins was affected byEETs in an antiapoptotic manner, defining a mechanism forthe observed protection of cardiomyocytes after injury byH/R. EETs may stimulate other pathways in the cell thatalso have a prosurvival role (e.g., opening of KATP chan-
nels). Future studies are needed to address whether theactivation of PI3K regulates channel function or whethermultiple prosurvival pathways must be functional at thesame time to protect cardiomyocytes. Understanding clearlyhow EETs are protective will be a first step toward devel-
Fig. 8. EET increases intracellular pAkt. HL-1 cells ( A) and neonatal myocytes ( B) were subjected to H/R after pretreatment with vehicle or 14,15-EET. Cellswere treated as described in MATERIALS AND METHODS and stained with pAkt antibody. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole, and thecells were viewed under fluorescent microscopy. Images were captured and overlaid as shown. C and D represent the quantitation of green fluorescence, whichindicates the increase in pAkt levels. *P 0.05 compared with control; #P 0.05 compared with H/R.
H731PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org

7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 11/14
oping these fatty acids into therapeutic agents for cardio-vascular disease.
The PI3K/Akt pathway is one of the most potent intracellu-lar mechanisms to promote cell survival. For example, in astudy of four downstream effectors of growth factor receptors,PI3K, Ras, Raf, and Src, PI3K was the only one to inhibitapoptosis after serum withdrawal (30). Our results indicate thatPI3K activity is enhanced by an EET within 30 min of application and also hours later during the first hour of reoxy-
genation. It is not clear whether EETs directly stimulate PI3Kor act indirectly. The best known activators of PI3K include Gprotein-coupled receptors (GPCRs) (10, 35, 40, 51, 69), recep-tor tyrosine kinases (23, 33), and glycoprotein 130 (12, 32, 56).Heterotrimeric G proteins and receptor tyrosine kinases medi-ate the action of EET. Evidence of cholera toxin-sensitive,high-affinity binding of EETs to mononuclear cells (61, 62) orthe activation of K channels via the stimulatory G protein-(Gs) subunit of heterotrimeric G proteins raises the possibilitythat EETs may stimulate PI3K via GPCR. The sulfonamidederivative of 14,15-EET (20 M 14,15-EET-SI) induced the
association between the EGF receptor and Src-like kinaseswithin 1 min of application at a concentration of 20 M inrenal epithelial cells (7). This high concentration of 14,15-EET-SI stimulated the phosphorylation of the 85-kDa regula-tory subunit of PI3K (7). Cross talk with the EGF receptorregulates the angiogenic action of EET in the chick cho-rioallantoic vessels (38). In addition, EETs can transcription-ally upregulate proteins such as the tissue plasminogen activa-tor via Gs (45), implying that secondary or late signaling may
occur after the synthesis of new proteins or factors that areinduced by EET. These observations may explain why a singleexposure to free fatty acids, which are rapidly metabolized incells, may trigger events appearing many hours later.
One downstream target of PI3K is the kinase Akt (67),which is reported to be activated during the intracellular signaltransduction of many receptors and survival factors. Membersof this family include Akt1, Akt2, and Akt3 (42). Akt, alsoknown as protein kinase B or RAC-PK (related to A and Ckinases), is a serine-threonine kinase. PI3K phosphorylatesinositol lipids that recruit and modify several targets containing
Fig. 9. Western blot analysis demonstratingeffect of EETs on Akt, Bcl-xl/Bcl-2-associateddeath promoter (BAD), and X-linked inhibitorof apoptosis (XIAP). HL-1 cells and neonatalmyocytes were grown and subjected to HRwith or without pretreatment with EETs. Thecells were lysed in the presence of phosphataseinhibitors, and the lysates were developed byWestern blot analysis with antibody to pAktand Akt ( A); samples with WT (0.2–1 M)were pretreated for 30 min. S-473, serine 473.
B: pBAD, BAD, and XIAP in neonatal myo-cytes (left ) and HL-1 cells (right ). Samplestreated with EET showed increase in phosphor-ylation of Akt (pAkt) and BAD (pBAD) andhigher levels of XIAP in both cell types,whereas Akt and BAD remain unaltered anddemonstrate equal protein loading in all lanesin A and B, respectively.
Fig. 10. EETs decrease caspase-9 activity.HL-1 cells ( A) and neonatal myocytes ( B) weresubjected to HR after pretreatment with vehicleor the 3 regioisomers of EET. Caspase-9 activ-ity was measured as described in MATERIALS
AND METHODS. *P 0.05, significant differencecompared with control (8 h); #P 0.05, sig-nificant difference compared with control (16h); &P 0.05, significant difference comparedwith HR (8 h); $P 0.05, significant differ-ence compared with HR (16 h). Controls weretreated under normoxic conditions. Both celltypes showed increased caspase-9 activity afterHR, which was attenuated by 8,9-, 11,12-, or14,15-EET by 16 h.
H732 PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org
7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 12/14
Pleckstrin homology (PH) domains including Akt and 3-phos-phoinositide-dependent kinase-1 (PDK-1) (4, 22, 31). Akt1 isactivated by phosphorylation first on Thr308 by PDK-1 andsubsequently on Ser473 (53). PDK-1 has a PH domain and is,therefore, also recruited to the membrane after the activation of PI3K. A number of downstream targets that regulate apoptosisare modulated by pAkt including members of the Bcl-2/CED9family Bax, Bak, and BAD (14, 26, 63, 68), XIAP, caspase-9,GSK-3, transcription factors of the forkhead, NF-B families,and endothelial nitric oxide synthase (18, 34, 49, 59). BAD andcaspase-9 are inactivated after phosphorylation by Akt (1),whereas XIAP is stabilized by it. Therefore, Akt promotes cellsurvival by ultimately modifying the core death machinery.
Akt primarily triggers the phosphorylation of BAD at Ser136,which is sufficient to promote survival. The phosphorylation of
BAD leads to the prevention of cell death via a mechanism thatinvolves the selective association of pBAD with isoforms of the scaffold protein 14–3-3. This interaction induces the re-lease of antiapoptotic-binding partners of BAD such as Bcl-xL
or Bcl-2 (14, 24, 65). 14–3-3 isoforms associate with a numberof cellular signaling molecules including the kinase suppressorof Ras, cdc25, Raf-1, and PI3K (3, 39). Growth factor depri-vation or apoptotic signals cause a removal of the phosphates,which allow BAD to bind to antiapoptotic Bcl-2, ultimatelyresulting in the displacement of voltage-dependent anion chan-nel-2 of the mitochondria, increasing mitochondrial outermembrane permeabilization and inducing apoptosis. BAD is,therefore, a proapoptotic molecule when it is not phosphory-lated.
XIAP is, as its name suggests, an inhibitor of apoptosis thatbelongs to the inhibitor of apoptosis family of proteins with anevolutionarily conserved role in regulating programmed celldeath (15, 16). It binds to and blocks caspases-9, -3, and -7
(16). The protein contains baculovirus inhibitors of apoptosisrepeats, which limit substrate access to catalytic sites of acti-vated caspases. In addition, this protein prevents the dimeriza-tion of procaspase-9 (57) and protects the cells from accidentalactivation of caspases. XIAP is an important antiapoptoticprotein since it attenuates the major inhibitor of the extrinsicpathway, caspase-9, as well as the common mediator of boththe intrinsic as well as extrinsic pathways, caspase-3 (13). Ourresults (Fig. 9) demonstrate reduced levels of this protein afterH/R in both types of cultured myocytes. Pretreatment withEETs impressively reduces this fall in the level of XIAPinduced by H/R.
Although this is the first report describing antiapoptoticactions of EETs on cardiomyocytes, the enhanced survival of
other cultured cells by these fatty acids and also by epoxy-genase overexpression has been reported. Examples of celltypes so affected include porcine kidney proximal tubule-likeepithelial cell line (LLCPKc14) (6), primary human coronaryand pulmonary microvascular endothelial cells (17), bovineaortic endothelial cells (66), and the human carcinoma cell lineTca-8,113 (29). The PI3K/Akt pathway has been implicated inall of these studies, although much higher concentrations of 14,15-EET (10 –20 M) were used in the kidney cells (6),where Akt kinase activity was increased 10 min after theaddition of 14,15-EET to the LLCPKc14s. Levels of pAkt werehigher after at least 12 h of treatment of the carcinoma cellswith 8,9-, 11,12-, or 14,15-EET (100 nM), followed by the
Fig. 11. EET protects contractility of primaryneonatal myocytes in culture. Neonatal myo-cytes were subjected to HR after pretreatmentwith vehicle or 14,15-EET (1 M). The numberof beating cells in a microscope field ( A) and thefrequency (beats/min; B) were counted as de-scribed in MATERIALS AND METHODS (a total of
3–5 batches of cells were examined for eachtreatment). Pretreatment with EET increasedboth total number and frequency of beating cellscompared with myocytes that were exposed toHR in the presence of vehicle. *P 0.05,significant difference compared with control;#P 0.05, significant difference comparedwith HR.
Fig. 12. Schematic representation of a pathway for protection of cardiomyo-cytes by EET via the PI3K/Akt prosurvival pathway. We have reported thatpretreatment with EET increases 1) PI3K activity, 2) phosphorylation of Akt,3) phosphorylation of BAD, 4) intracellular XIAP, and 5) inhibition of activityof caspases-9. Each of these events decreases activity of caspase-3, a lateeffector, to protect the cardiomyocyte from apoptosis by HR. These targets areknown to align in a signaling cascade as illustrated in the schematic. It shouldbe noted that the results do not implicate absence of other prosurvival ormitotic pathways that may also be targeted by EETs. RTK, receptor tyrosinekinases.
H733PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org

7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 13/14
induction of apoptosis by tumor necrosis factor- for 12 h (29).Interestingly, the expression of the 110-kDa PI3K subunit wasincreased after the overexpression of epoxygenase enzymesthat catalyze formation of EET in bovine aortic endothelialcells (66).
Hence, we demonstrate that EETs prevent apoptosis in ratneonatal cardiac myocytes and a mouse atrial cardiomyocyte
cell line (HL-1) subjected to H/R injury. We report for the firsttime that treatment with EET enhances PI3K activity (Fig. 7).This treatment also attenuates both the increase in activity of caspase-9 and the fall in the levels of intracellular XIAPinduced by H/R. We have confirmed that EETs increasephosphorylation of Akt that is upstream of these effectors.Together, these observations support mechanistic evidence forthe protective effect of EETs in cardiomyocytes to preventH/R-induced cell death. These data make it essential to inves-tigate whether the activation of potassium channels (especiallyKATP channels), the mechanism favored by other investigatorsfor the protection of myocardium by EETs, is independent of PI3K or occurs by cross talk in parallel to this prosurvivalpathway.
ACKNOWLEDGMENTS
Technical help from Laurel Dunn and Ying Gao are gratefully acknowl-edged.
GRANTS
Financial support was provided by National Heart, Lung, and BloodInstitute Grants HL-069996 (to M. Medhora), HL-49294 (to E. R. Jacobs), andHL-68627 (to E. R. Jacobs) and the Robert A. Welch Foundation GrantGM-31278 (to J. R. Falck).
REFERENCES
1. Amaravadi R, Thompson CB. The survival kinases Akt and Pim aspotential pharmacological targets. J Clin Invest 115: 2618–2624, 2005.
2. Bialik S, Geenen DL, Sasson IE, Cheng R, Horner JW, Evans SM,
Lord EM, Koch CJ, Kitsis RN. Myocyte apoptosis during acute myo-cardial infarction in the mouse localizes to hypoxic regions but occursindependently of p53. J Clin Invest 100: 1363–1372, 1997.
3. Burbelo PD, Hall A. 14-3-3 proteins. Hot numbers in signal transduction.Curr Biol 5: 95–96, 1995.
4. Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylino-sitol-3-OH kinase signal transduction. Nature 376: 599–602, 1995.
5. Campbell WB, Harder DR. Endothelium-derived hyperpolarizing fac-tors and vascular cytochrome P450 metabolites of arachidonic acid in theregulation of tone. Circ Res 84: 484–488, 1999.
6. Chen JK, Capdevila J, Harris RC. Cytochrome p450 epoxygenasemetabolism of arachidonic acid inhibits apoptosis. Mol Cell Biol 21:6322–6331, 2001.
7. Chen JK, Falck JR, Reddy KM, Capdevila J, Harris RC. Epoxyeico-satrienoic acids and their sulfonimide derivatives stimulate tyrosine phos-
phorylation and induce mitogenesis in renal epithelial cells. J Biol Chem273: 29254–29261, 1998.8. Chen YJ, Jiang H, Quilley J. The nitric oxide and prostaglandin-
independent component of the renal vasodilator effect of thimerosal ismediated by epoxyeicosatrienoic acids. J Pharmacol Exp Ther 304:1292–1298, 2003.
9. Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury intransgenic mice. Am J Physiol Heart Circ Physiol 280: H2313–H2320,2001.
10. Chesley A, Lundberg MS, Asai T, Xiao RP, Ohtani S, Lakatta EG,Crow MT. The 2-adrenergic receptor delivers an antiapoptotic signal tocardiac myocytes through Gi-dependent coupling to phosphatidylinositol3-kinase. Circ Res 87: 1172–1179, 2000.
11. Claycomb WC, Lanson NA Jr, Stallworth BS, Egeland DB, DelcarpioJB, Bahinski A, Izzo NJ Jr. HL-1 cells: a cardiac muscle cell line that
contracts and retains phenotypic characteristics of the adult cardiomyo-cyte. Proc Natl Acad Sci USA 95: 2979–2984, 1998.
12. Craig R, Wagner M, McCardle T, Craig AG, Glembotski CC. Thecytoprotective effects of the glycoprotein 130 receptor-coupled cytokine,cardiotrophin-1, require activation of NF-kappa B. J Biol Chem 276:37621–37629, 2001.
13. Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial deathpathway and cardiac myocyte apoptosis. Circ Res 95: 957–970, 2004.
14. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, GreenbergME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241, 1997.
15. Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srini-
vasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptoticevents induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 17: 2215–2223, 1998.
16. Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is adirect inhibitor of cell-death proteases. Nature 388: 300–304, 1997.
17. Dhanasekaran A, Al-Saghir R, Lopez B, Zhu D, Guttermann DD,
Jacobs ER, Medhora M. Protective effects of epoxyeicosatrienoic acidson human endothelial cells from the pulmonary and coronary vasculature.
Am J Physiol Heart Circ Physiol 291: H517–H531, 2006.18. Downward J. PI3-kinase, Akt and cell survival. Semin Cell Dev Biol 15:
177–182, 2004.19. Fleming I. Cytochrome P450 and vascular homeostasis. Circ Res 89:
753–762, 2001.
20. Fleming I, Michaelis UR, Bredenkotter D, Fisslthaler B, Dehghani F,Brandes RP, Busse R. Endothelium-derived hyperpolarizing factor syn-thase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res 88: 44–51, 2001.
21. Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocar-dium. Circ Res 79: 949–956, 1996.
22. Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK,
Kaplan DR, Tsichilis PN. The protein kinase encoded by the Aktproto-oncogene is a target of the PDGF-activated phosphatidylinositol3-kinase. Cell 81: 727–736, 1995.
23. Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotessurvival of cardiomyocytes in vitro and protects against ischemia-reper-fusion injury in mouse heart. Circulation 101: 660–667, 2000.
24. Gajewski TF, Thompson CB. Apoptosis meets signal transduction:elimination of a BAD influence. Cell 87: 589–592, 1996.
25. Gebremedhin D, Harder DR, Pratt PF, Campbell WB. Bioassay of an
endothelium-derived hyperpolarizing factor from bovine coronary arteries:role of a cytochrome P450 metabolite. J Vasc Res 35: 274–284, 1998.
26. Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N.
Inhibition of early apoptotic events by Akt/PKB is dependent on the firstcommitted step of glycolysis and mitochondrial hexokinase. Genes Dev
15: 1406–1418, 2001.27. Gross GJ, Hsu A, Falck JR, Nithipatikom K. Mechanisms by which
epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J
Mol Cell Cardiol 42: 687–691, 2007.28. Guerra S, Leri A, Wang X, Finato N, Di Loreto C, Beltrami CA,
Kajstura J, Anversa P. Myocyte death in the failing human heart isgender dependent. Circ Res 85: 856–866, 1999.
29. Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, Ning YG,
Xiao X, Zeldin DC, Wang DW. Cytochrome P450 2J2 promotes theneoplastic phenotype of carcinoma cells and is up-regulated in humantumors. Cancer Res 65: 4707–4715, 2005.
30. Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis
PN, Hay N. The PI3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev 11: 701–713, 1997.
31. Klarlund JK, Guilherme A, Holik JJ, Virbasius JV, Chawla A, Czech
MP. Signaling by phosphoinositide-3,4,5-trisphosphate through proteinscontaining pleckstrin and Sec7 homology domains. Science 275: 1927–1930, 1997.
32. Kuwahara K, Saito Y, Kishimoto I, Miyamoto Y, Harada M, Ogawa
E, Hamanaka I, Kajiyama N, Takahashi N, Izumi T, Kawakami R,
Nakao K. Cardiotrophin-1 phosphorylates Akt and BAD, and prolongscell survival via a PI3K-dependent pathway in cardiac myocytes. J Mol
Cell Cardiol 32: 1385–1394, 2000.33. Matsui T, Li L, del Monte F, Fukui Y, Franke TF, Hajjar RJ,
Rosenzweig A. Adenoviral gene transfer of activated phosphatidylinositol3-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro.Circulation 100: 2373–2379, 1999.
H734 PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt
AJP-Heart Circ Physiol • VOL 294 • FEBRUARY 2008 • www.ajpheart.org

7/31/2019 Anu Akt Apoptosis
http://slidepdf.com/reader/full/anu-akt-apoptosis 14/14
34. Matsui T, Rosenzweig A. Convergent signal transduction pathwayscontrolling cardiomyocyte survival and function: the role of PI3-kinaseand Akt. J Mol Cell Cardiol 38: 63–71, 2005.
35. Means CK, Xiao CY, Li Z, Zhang T, Omens JH, Ishii I, Chun J,Brown JH. Sphingosine1-phosphate S1P2 and S1P3 receptor-mediatedAkt activation protects against in vivo myocardial ischemia-reperfusioninjury. Am J Physiol Heart Circ Physiol 292: H2944–H2951, 2007.
36. Medhora M, Dhanasekaran A, Gruenloh SK, Dunn LK, GabrilovichM, Falck JR, Harder DR, Jacobs ER, Pratt PF. Emerging mechanismsfor growth and protection of the vasculature by cytochrome P450-derivedproducts of arachidonic acid and other eicosanoids. Prostaglandins Other
Lipid Mediat 82: 19–29, 2007.37. Miao W, Luo Z, Kitsis RN, Walsh K. Intracoronary, adenovirus-
mediated Akt gene transfer in heart limits infarct size following ischemia-reperfusion injury in vivo. J Mol Cell Cardiol 32: 2397–2402, 2000.
38. Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse
R. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induceangiogenesis via cross-talk with the epidermal growth factor receptor(EGFR). FASEB J 17: 770–772, 2003.
39. Morrison D. 14-3-3: modulators of signaling proteins? Science 266:56–57, 1994.
40. Naga Prasad SV, Esposito G, Mao L, Koch WJ, Rockman HA. Gbetagamma-dependent phosphoinositide 3-kinase activation in hearts within vivo pressure overload hypertrophy. J Biol Chem 275: 4693–4698,2000.
41. Natarajan R, Reddy MA. HETEs/EETs in renal glomerular and epithe-lial cell functions. Curr Opin Pharmacol 3: 198–203, 2003.42. Nicholson KM, Anderson NG. The protein kinase B/Akt signaling
pathway in human malignancy. Cell Signal 14: 381–395, 2002.43. Nithipatikom K, Moore JM, Isbell MA, Falck JR, Gross GJ. Epoxyei-
cosatrienoic acids (EETs) in cardioprotection: ischemic versus reperfusioninjury. Am J Physiol Heart Circ Physiol 291: H537–H542, 2006.
44. Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, LiaoJK. Anti-inflammatory properties of cytochrome P450 epoxygenase-de-rived eicosanoids. Science 285: 1276–1279, 1999.
45. Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, Liao JK.Activation of Galpha s mediates induction of tissue-type plasminogenactivator gene transcription by epoxyeicosatrienoic acids. J Biol Chem276: 15983–15989, 2001.
46. Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA,Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa
P. Apoptosis in the failing human heart. N Engl J Med 336: 1131–1141,
1997.47. Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E,
Gambert SR, Cigola E, Anversa P. Acute myocardial infarction inhumans is associated with activation of programmed myocyte cell death inthe surviving portion of the heart. J Mol Cell Cardiol 28: 2005–2016,1996.
48. Oltman CL, Weintraub NL, VanRollins M, Dellsperger KC. Epoxyei-cosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodila-tors in the canine coronary microcirculation. Circ Res 83: 932–939, 1998.
49. Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, BackxPH. The role of phosphoinositide-3 kinase and PTEN in cardiovascularphysiology and disease. J Mol Cell Cardiol 37: 449–471, 2004.
50. Palojoki E, Saraste A, Eriksson A, Pulkki K, Kallajoki M, Voipio-Pulkki LM, Tikanen I. Cardiomyocyte apoptosis and ventricular remod-eling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol280: H2726–H2731, 2001.
51. Robert P, Tsui P, Laville MP, Livi GP, Sarau HM, Bril A, Berrebi-
Bertrand I. EDG1 receptor stimulation leads to cardiac hypertrophy in ratneonatal myocytes. J Mol Cell Cardiol 33: 1589–1606, 2001.
52. Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82: 131–185, 2002.
53. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation andregulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098–1101, 2005.
54. Seubert JM, Sinal CJ, Graves J, DeGraff LM, Bradbury JA, Lee CR,Goralski K, Carey MA, Luria A, Newman JW, Hammock BD, FalckJR, Roberts H, Rockman HA, Murphy E, Zeldin DC. Role of solubleepoxide hydrolase in postischemic recovery of heart contractile function.Circ Res 99: 442–450, 2006.
55. Seubert JM, Zeldin DC, Nithipatikom K, Gross GJ. Role of epoxyei-cosatrienoic acids in protecting the myocardium following ischemia/ reperfusion injury. Prostaglandins Other Lipid Mediat 82: 50–59, 2007.
56. Sheng Z, Knowlton K, Chen J, Hoshijima M, Brown JH, Chien KR.Cardiotrophin 1 (CT-1) inhibition of cardiac myocyte apoptosis via amitogen-activated protein kinase-dependent pathway. Divergence fromdownstream CT-1 signals for myocardial cell hypertrophy. J Biol Chem272: 5783–5791, 1997.
57. Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM,Alnemri ES, Fairman R, Shi Y. Mechanism of XIAP-mediated inhibi-tion of caspase-9. Mol Cell 11: 519–527, 2003.
58. Sun J, Sui X, Bradbury JA, Zeldin DC, Conte MS, Liao JK. Inhibitionof vascular smooth muscle cell migration by cytochrome p450 epoxyge-nase-derived eicosanoids. Circ Res 90: 1020–1027, 2002.
59. Wang H, Lin L, Jiang J, Wang Y, Lu ZY, Bradbury JA, Lih FB,Wang DW, Zeldin DC. Up-regulation of endothelial nitric-oxide synthaseby endothelium-derived hyperpolarizing factor involves mitogen-activated
protein kinase and protein kinase C signaling pathways. J Pharmacol ExpTher 307: 753–764, 2003.60. Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor
SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiacmyocyte apoptosis in heart failure. J Clin Invest 111: 1497–1504, 2003.
61. Wong PY, Lai PS, Shen SY, Belosludtsev YY, Falck JR. Post-receptorsignal transduction and regulation of 14( R),15(S)-epoxyeicosatrienoic acid(14,15-EET) binding in U-937 cells. J Lipid Mediat Cell Signal 16:155–169, 1997.
62. Wong PY, Lin KT, Yan YT, Ahern D, Iles J, Shen SY, Bhatt RK,
Falck JR. 14( R),15(S)-epoxyeicosatrienoic acid (14,15-EET) receptor inguinea pig mononuclear cell membranes. J Lipid Mediators 6: 199–208,1993.
63. Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cellsurvival and apoptosis by inhibiting Bax conformational change. Onco-
gene 20: 7779–7786, 2001.64. Yang B, Graham L, Dikalov S, Mason RP, Falck JR, Liao JK, Zeldin
DC. Overexpression of cytochrome P450 CYP2J2 protects against hyp-oxia-reoxygenation injury in cultured bovine aortic endothelial cells . MolPharmacol 60: 310–320, 2001.
65. Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ.Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax andpromotes cell death. Cell 80: 285–291, 1995.
66. Yang S, Lin L, Chen JX, Lee CR, Seubert JM, Wang Y, Wang H,Chao ZR, Tao DD, Gong JP, Lu ZY, Wang DW, Zeldin DC. Cyto-chrome P-450 epoxygenases protect endothelial cells from apoptosisinduced by tumor necrosis factor- via MAPK and PI3K/Akt signalingpathways. Am J Physiol Heart Circ Physiol 293: H142–H151, 2007.
67. Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in theprevention of apoptosis by nerve growth factor. Science 267: 2003–2006,1998.
68. Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphor-ylation of death agonist BAD in response to survival factor results inbinding to 14-3-3 not BCL-X(l). Cell 87: 619–628, 1996.
69. Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP.Dual modulation of cell survival and cell death by 2-adrenergic signalingin adult mouse cardiac myocytes. Proc Natl Acad Sci USA 98: 1607–1612,2001.
H735PROTECTION OF CARDIOMYOCYTES BY EETs VIA PI3K/Akt