28700 san sebastiÁn de los reyes (madrid) espaÑa nº ...intranet2.ciboma.org/2004-01/protocolo...
TRANSCRIPT

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 1 de 82
COALICIÓN IBEROAMERICANA DE INVESTIGACIÓN EN ONCOLOGÍA MAMARIA (CIBOMA)
CIBOMA/2004-01
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA
DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE,
RECEPTORES HORMONALES Y HER2neu NEGATIVOS CIBOMA/2004-01
Quimioterapia vs. observación PROMOTOR: CIBOMA (COALICIÓN IBEROAMERICANA DE INVESTIGACIÓN EN ONCOLOGÍA MAMARIA) AVENIDA DE LOS PIRINEOS 7, OFICINA 1-14 28700 SAN SEBASTIÁN DE LOS REYES (MADRID) ESPAÑA Nº EudraCT: 2005-002838-36 COORDINADORES MÉDICOS DEL ESTUDIO Dra. Ana Lluch Servicio de Oncología Medica Hospital Clínico U. De Valencia Av. Blasco Ibáñez 17 46010 Valencia (España) Tel:+34963862625 Fax:+34963622238
Dra. Laura Torrecillas Centro Médico de Especialidades 20 de Noviembre ISSSTE México Tel: +52-55-5575 3072 Fax:: +52-55-5575 3072
Dr. Carlos H. Barrios Servicio de Oncología Médica Hospital Sao Lucas-PUC Avda. Iìranga, 6690 SL 228-Porto Alegre RS CEP 90,610-000 Brasil Tel: + 55 51 33203319 Fax: + 55 51 33203319
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL: La información y los datos incluidos en este protocolo contienen información confidencial que es propiedad de la Coalición Iberoamericana de Investigación en Oncología Mamaria (CIBOMA). Ninguna persona está autorizada a hacerla pública sin previo permiso por escrito de CIBOMA. Estas limitaciones se aplicarán, igualmente, a toda la información considerada como privilegiada o confidencial que se le facilite en el futuro. Este material podrá ser divulgado y utilizado por su equipo y colaboradores, según sea necesario para la realización del estudio clínico
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 2 de 82
1.- RESUMEN DEL ESTUDIO Tipo de solicitud Ensayo clínico de una especialidad farmacéutica en una nueva indicación. Identificación del promotor
Coalición Iberoamericana de Investigación en Oncología Mamaria (CIBOMA) Avenida de los Pirineos 7, oficina 1-14 28700 San Sebastián de los Reyes (Madrid) España Tel.: + 34 916592870 Fax: +34 916510406 Correo electrónico: [email protected] Página web: www.ciboma.org
Título del estudio “Estudio fase IV.III multicéntrico, abierto, de asignación aleatoria de tratamiento, para evaluar la eficacia de terapia de mantenimiento con capecitabina (X) tras quimioterapia adyuvante estándar en pacientes con cáncer de mama operable, receptores hormonales y HER2neu negativos”.
Código del protocolo
CIBOMA/2004-01
Investigadores principales
Dra. Ana Lluch, Dr. Ruiz Borrego, Dra. Calvo (España), Dra. Laura Torrecillas (México), Dr. Carlos H. Barrios (Brasil) (véase apéndice 6).
Centros en los que se prevé realizar el ensayo
Hospital Clínico Universitario de Valencia, Hospital Universitario Virgen del Rocío, Complejo Hospitalario Juan Canalejo (España). Centro Médico de Especialidades 20 de Noviembre (México). Hospital Sao Lucas-PUC (Brasil) (véase apéndice 6).
Comités Éticos de Investigación Clínica que han aprobado el ensayo
Los correspondientes a los centros participantes (véase apéndice 6). España: el Comité Ético de Investigación Clínica (CEIC) que actúa como CEIC de referencia para el dictamen único es el Comité Ético de Investigación Clínica de Galicia.
Monitores de Buena Práctica Clínica (BPC) del estudio
Andrés Hernando: [email protected] Gema Monzón: gmonzó[email protected] Yago Parga: [email protected] Yolanda Amigo: [email protected] Ruth Campo de la Fuente: [email protected] Victoria Ruiz: [email protected]
Coordinación global del proyecto
Dra. Esther Mahillo CIBOMA Avenida de los Pirineos 7, oficina 1-14 28700 San Sebastián de los Reyes (Madrid) España Tel: +34916592870 Fax:+34916510406 Correo electrónico: [email protected]
Coordinación administrativa
Coordinación administrativa del proyecto: Elena Gutiérrez/María del Carmen Pastor CIBOMA Avenida de los Pirineos 7, oficina 1-14 28700 San Sebastián de los Reyes (Madrid) España Tel: +34916592870 Fax:+34916510406 Correo electrónico: [email protected]; [email protected]

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 3 de 82
Equipo estadístico Mª José Escudero y María Isabel Casas CIBOMA Avenida de los Pirineos 7, oficina 1-14 28700 San Sebastián de los Reyes (Madrid) España Tel: +34916592870 Fax:+34916510406 Correo electrónico: [email protected]; [email protected]
Coordinador de gestión de datos
César García CIBOMA Avenida de los Pirineos 7, oficina 1-14 28700 San Sebastián de los Reyes (Madrid) España Tel: +34916592870 Fax:+34916510406 Correo electrónico: [email protected]
Laboratorio central de análisis de muestras patológicas
Dr. José Schneider Universidad Rey Juan Carlos, Facultad de Ciencias de la Salud Departamento de Ciencias de la Salud-II Laboratorio de la Unidad Materno-Infantil, despacho 1.024. Avda de Atenas, S/N, 28922 Alcorcón (Madrid) España Teléfono: +34 91 488 8888.
Fármaco experimental y control
Brazo control: Observación Brazo experimental: capecitabina 1000 mg/ m2 dos veces al día durante 14 días, seguidos de un periodo de descanso de 7 días, durante 8 ciclos.
Fase de ensayo clínico
Se trata de un ensayo clínico en fase IV.III. Es fase IV porque se utiliza un producto en investigación que cuenta con una licencia de comercialización en todos los países participantes. A la vez, el producto en investigación se va a administrar en una indicación diferente a la de la licencia de comercialización para evaluar su eficacia en términos de incremento de supervivencia libre de enfermedad (diseño de estudio fase III).
Objetivos Objetivo Primario: comparar la supervivencia libre de enfermedad a 5 años después de terapia de mantenimiento con 8 ciclos de capecitabina (X) frente a observación, en pacientes con cáncer de mama operable, receptores hormonales y HER2neu negativos, que han recibido un tratamiento quimioterápico adyuvante estándar. Objetivos secundarios:
?? Comparar la Supervivencia Global (SG) entre los dos grupos mencionados anteriormente.
?? Comparar la toxicidad entre los 2 grupos mencionados anteriormente. Objetivos terciarios Determinar el efecto del tratamiento con capecitabina sobre la aparición de amenorrea en mujeres premenopáusicas y duración de la misma. (En centros seleccionados): comprobar la existencia de polimorfismos de la enzima timidilato sintasa (TS) y en la enzima metilentetrahidrofolato reductasa (MTHFR), y confirmar su asociación con la toxicidad y eficacia del tratamiento con capecitabina.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 4 de 82
Diseño del estudio
Ensayo fase IV.III prospectivo, abierto, de asignación aleatoria de tratamiento. Las pacientes se estratificarán en el momento de la inclusión, después de completar al menos 6 ciclos de quimioterapia adyuvante con antraciclinas y/o taxanos, según el hospital participante, el tratamiento adyuvante previo (antraciclinas vs. antraciclinas y taxanos, y el número de ganglios axilares ipsilaterales afectados (0, 1-3, ?4), y se distribuirán de forma aleatoria para recibir: Xx8: capecitabina 1000 mg/m2 p.o. administrados 2 veces al día (mañana y noche)
durante 14 días, seguidos de un periodo de descanso de 7 días, durante 8 ciclos.
Observación Se ha planeado la reducción de la dosis y/o el aplazamiento del tratamiento y la suspensión del tratamiento en el caso de que se manifiesten toxicidades severas de tipo hematológico y no hematológico. Indicación para radioterapia -Los dos grupos: Las pacientes tratadas con tumorectomía se someterán a radioterapia postoperatoria después de finalizar la quimioterapia adyuvante estándar y de la resolución de cualquier efecto secundario. Se podrá administrar radioterapia después de la mastectomía e irradiar los ganglios ipsilaterales a discreción del radioterapeuta. Se recogerán las directrices de radioterapia de cada institución. Deberán transcurrir como máximo 4 semanas entre el final de la radioterapia y el registro de las pacientes en el estudio CIBOMA/2004-01. Estado de receptores de estrógeno y/o progesterona: Se deberá realizar un análisis en el laboratorio central designado, del estado de los receptores de estrógeno y progesterona en una muestra del tumor primario de las pacientes. Se deberán conocer los resultados antes de la randomización. Aquellos tumores que no puedan considerarse definitivamente negativos, es decir, los limítrofes, se considerarán como positivos. Estado de expresión de HER2: se deberá realizar un análisis por inmunohistoquímica en el laboratorio central designado, del estado de expresión de la proteina HER2. En los tumores con un resultado de 2+, será necesario determinar el número de copias del gen c-erbB2, por FISH, en la muestra de tumor primario de las pacientes. Perfil de genotipo basal: se deberá realizar un análisis por inmunohistoquímica en el laboratorio central designado de las citoqueratinas CK 5/6 y del estado de expresión del receptor del EGF (REGF).
Enfermedad o trastorno en estudio
Pacientes con cáncer de mama operado y sin afectación metastásica (AJCC, 2002). Las pacientes podrán participar tanto si presentan (ganglios positivos) como si no (ganglios negativos) afectación axilar ganglionar. Las pacientes con ganglios negativos deberán presentar un tumor de al menos 2 cm de diámetro. El numero de ganglios axilares estudiados no deberá ser inferior a 6. En el caso de pacientes sometidas a biopsia del ganglio centinela, el mismo contará a los efectos de número de ganglios extraídos y afectados por la enfermedad. Los tumores de las pacientes deberán presentar un estado negativo de los receptores hormonales de estrógeno y progesterona, y de HER2, mediante determinación en laboratorio central designado.
Variable principal de valoración
Supervivencia libre de enfermedad a los 5 años.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 5 de 82
Criterios de inclusión
1. Se debe obtener y documentar el consentimiento informado por escrito antes de iniciar los procedimientos específicos del protocolo, incluida la cooperación esperada de las pacientes en el tratamiento y el seguimiento, de acuerdo con las normas ICH de Buena Práctica Clínica. La paciente deberá firmar un consentimiento informado para envío de su muestra tumoral a laboratorio central. Posteriormente, si la paciente es elegible, deberá firmar un consentimiento para su participación en el ensayo clínico.
2. Cáncer de mama histológicamente demostrado (examen histológico del tumor: adenocarcinoma invasivo).
3. Pacientes con afectación axilar ganglionar ipsilateral. En caso de utilizarse la técnica del ganglio centinela, el mismo contará como ganglio extirpado y afectado. Se admitirán las pacientes que se puedan clasificar en los siguientes grupos (AJCC, 2002):
- pN1a: Metástasis en 1 a 3 ganglios axilares. - pN2a: Metástasis en 4 a 9 ganglios axilares (al menos un depósito
tumoral > 0,2 cm). - pN3a: Metástasis en 10 ó más ganglios axilares (al menos un
depósito tumoral > 0,2 cm). No serían elegibles las pacientes con diagnóstico pN3a con metástasis en ganglios infraclaviculares.
Nota: Pacientes sin afectación ganglionar axilar (N0) son también elegibles, siempre que el tumor primario mida más de 2 cm de diámetro. En caso de utilizarse la técnica del ganglio centinela, no será necesario realizar linfadenectomía. 4. Mujeres en régimen ambulatorio de edad? 18 años. 5. Índice del estado funcional de Karnofsky ? 80 (ECOG 0,1).
Criterios de exclusión
1. La paciente no ha sido sometida a tratamiento quirúrgico definitivo para el cáncer de mama operable (T1-3, M0) bien mastectomía, o cirugía conservadora de la mama; o los márgenes de la muestra extraída de la cirugía definitiva son histológicamente positivos para adenocarcinoma invasivo o carcinoma ductal in situ (DCIS). El carcinoma lobular in situ no se considera un margen positivo.
2. La paciente no ha recibido al menos 6 ciclos de quimioterapia adyuvante previa con esquemas que contengan antraciclinas y/o taxanos (docetaxel o paclitaxel). Los tratamientos permitidos se encuentran disponibles en el apéndice 1. Cualquier otro esquema de tratamiento deberá ser aprobado por los coordinadores médicos del estudio.
3. En la linfadenectomía, extirpación de menos de 6 ganglios (para las pacientes con biopsia del ganglio centinela, si este es positivo, cuenta como extirpado).
4. Terapia anterior con antraciclinas o taxanos (paclitaxel, docetaxel) para cualquier neoplasia diferente del cáncer de mama que está siendo tratado.
5. Para pacientes que no reciben radioterapia, el intervalo entre el día 1 del último ciclo de quimioterapia adyuvante y la entrada en el ensayo clínico es superior a 8 semanas (2 meses). Para las pacientes en las que está indicada la administración de radioterapia adyuvante, el intervalo entre la última sesión y la entrada en el estudio es superior a 4 semanas (1 mes).
6. Pacientes con tumores con receptores de estrógeno, progesterona y HER2 desconocidos, o positivos.
7. Pacientes embarazadas o en período de lactancia. Pacientes con potencial de fertilidad y un resultado desconocido o positivo en la prueba de embarazo en orina o suero, realizada durante los 14 días anteriores a la asignación de tratamiento.
8. Mujeres en edad fértil que no desean usar un método anticonceptivo fiable y apropiado. Las mujeres postmenopáusicas deben haber permanecido

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 6 de 82
amenorreicas durante al menos 12 meses para ser consideradas como no fértiles.
9. Diagnóstico de cáncer de mama bilateral invasivo. 10. Enfermedad cardiaca clínicamente relevante, tal como insuficiencia cardiaca
congestiva o enfermedad arterial coronaria sintomática, arritmias cardiacas no controladas con tratamiento o historia anterior de infarto de miocardio durante los 12 meses anteriores a la inclusión en el estudio, o hipertensión no controlada.
11. Historia de trastornos neurológicos o psiquiátricos significativos, incluidos trastornos sicóticos, demencia o ataques que impedirían a la paciente entender y otorgar el consentimiento informado, interferirían en el cumplimiento de la pauta del fármaco en estudio.
12. Infección activa no controlada , u otras enfermedades o patologías médicas graves, tales como ulcera péptica activa, diabetes mellitus inestable.
13. Presencia de anomalías en cualquiera de los parámetros de laboratorio, tal y como se definen a continuación: (en los 14 días anteriores a la asignación de tratamiento): ?? Hemoglobina < 10 mg/dl; recuento absoluto de neutrófilos < 1,5 x 109/l;
recuento de plaquetas < 100x 109/l. ?? ASAT (SGOT) y ALAT (SGPT) > 2,5 LSN; Fosfatasa alcalina > 5 LNS;
Bilirrubina total > 2.0 LSN. Las pacientes con valores de ASAT y/o ALAT > 1,5 x LNS asociados con un nivel de fosfatasa alcalina > 2,5 x LNS no se seleccionarán para el estudio.
?? Insuficiencia renal severa, definida como aclaramiento de creatinina calculado inferior a 30 ml/min, según la fórmula de Cockroft and Gault (véase apéndice 8) o creatinina sérica > 1,5 LSN.
14. Historia anterior o actual de neoplasias distintas al cáncer de mama, a excepción de:
?? Cáncer de piel no melanoma, carcinoma de cervix uterino in situ u otro tipo de tumor tratados de forma curativa y sin indicios de enfermedad durante, al menos, diez años.
?? Carcinoma ductal in situ (DCIS) ipsilateral de mama. ?? Carcinoma lobular in situ (LCIS) de mama.
15. Hipersensibilidad conocida a capecitabina, fluorouracilo o a cualquiera de los excipientes.
16. Pacientes con falta de integridad física del tracto gastrointestinal superior o que manifiesten síndrome de mala absorción, o imposibilidad de tomar medicación por vía oral.
17. Tratamiento previo con capecitabina o con infusión contínua (> 24 h) con 5-FU, o con otras fluoropirimidinas orales, tales como eniluracil/5-FU, uracilo/tegafur, S1 o emitefur.
18. Transfusiones de sangre o administración de factores de crecimiento para ayudar a la recuperación hematológica en las 2 semanas previas al comienzo del tratamiento del estudio.
19. Tratamiento hormonal en los 10 días previos al comienzo del tratamiento del estudio.
20. Posibilidad de reacción severa no prevista a terapia con fluoropirimidina, con o sin probada deficiencia de dihidropirimidina dehidrogenasa (DPD).
21. Transplantes alográficos de órganos que requieran terapia inmunosupresora. 22. Pacientes con tratamiento anticoagulante con derivados de cumarina. 23. Pacientes en tratamiento con sorivurina o sus análogos químicamente
relacionados, como la brivudina. 24. Tratamiento concomitante con otros fármacos en investigación. Participación
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 7 de 82
en otro ensayo clínico con cualquier fármaco en investigación no comercializado durante los 30 días previos a la inclusión en el estudio.
25. Tratamiento concomitante con cualquier otra terapia anticancerosa. 26. Pacientes no accesibles para el tratamiento y el seguimiento. Las pacientes
registradas en este ensayo deberán ser tratadas y seguidas en el centro participante, que podría ser el centro del investigador principal o co-investigador.
27. Pacientes que no puedan o quieran cumplir con el protocolo, considerando la duración completa del estudio.
Población en estudio y número total de pacientes
Se estima que la SLE a los 5 años en el brazo control es del 64,72%. Se pretende detectar una reducción del 25% en la razón de riesgos, con una potencia del 80% usando la prueba log-rank a un nivel bilateral del 0,05. En este caso, la SLE a 5 años en el brazo experimental sería del 72,2%. Esto requiere que el número de eventos para la SLE sea de 379 en total. Considerando un 10% de pérdidas post-randomización, el tamaño final será de 1324 pacientes, 662 por brazo de tratamiento El proceso de asignación aleatoria de tratamiento será centralizado y las pacientes se estratificarán según el centro participante, el tratamiento adyuvante previo (antraciclinas vs. antracicinas y taxanos), y el número de ganglios afectados (0 vs. 1-3 vs ? 4).
Duración del tratamiento
La duración previsible del tratamiento en la rama experimental con X es de 24 semanas. Se seguirá el mismo calendario de visitas para las pacientes en el brazo de observación. El periodo de reclutamiento durará 24 meses. Se estima un reclutamiento de 662 pacientes por año entre todos los países y centros participantes.
Calendario y fecha prevista de terminación
Inicio de la inclusión: enero de 2006. Fin de la inclusión: enero de 2008. Seguimiento clínico: 5 años. Análisis final: último trimestre de 2012.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 8 de 82
DISEÑO DEL ESTUDIO CIBOMA/2004-01: Se trata de un ensayo clínico prospectivo, de asignación aleatoria de tratamiento, para evaluar la eficacia y seguridad de terapia de mantenimiento con capecitabina, para mujeres con cáncer de mama operable, receptores hormonales y HER2neu negativos, que hayan recibido previamente un tratamiento adyuvante quimioterápico estándar. Las pacientes con ganglios negativos son elegibles si presentan un tamaño de tumor > 2 cm. El tratamiento control es la observación. El tratamiento experimental consiste en la administración de 8 ciclos de capecitabina a una dosis de 1000 mg/m2, dos veces al día, administrado p.o, durante 14 días, seguidos de una semana de descanso. El estudio se desarrollará siguiendo este esquema de eventos:
Descripción de la población en estudio. Pacientes con cáncer de mama operado sin afectación metastásica (AJCC, 2002). Serán elegibles las pacientes con afectación axilar ganglionar (ganglios positivos), o sin afectación ganglionar axilar (ganglios negativos), pero en este caso, el tumor deberá ser mayor de 2 cm de diámetro. El numero de ganglios axilares estudiados no deberá ser inferior a 6. En el caso de pacientes sometidas a biopsia del ganglio centinela, el mismo contará a los efectos de número de ganglios extraídos y afectados por la enfermedad. Sólo serán elegibles las pacientes con tumores con receptores de estrógeno y progesterona y HER2 negativos. Estado de receptores de estrógeno y/o progesterona: se deberá realizar un análisis de los receptores de estrógeno y/o progesterona en una muestra del tumor primario de las pacientes, mediante envío al laboratorio central designado. El mejor momento para el envío de la muestra se considera que es el inicio del tratamiento con la quimioterapia estándar. Aquellos tumores que no puedan considerarse definitivamente negativos, es decir, los limítrofes, se considerarán
Cáncer de mama operado RE/RP Negativos
HER2neu negativo T1-3, N+, M0 / T2-3, N0, M0
QT adyuvante estándar +/- RT complementaria
Distribución aleatoria: ?? Centro participante, ?? Tratamiento adyuvante previo (antraciclinas
vs. Antracicinas y taxanos), ?? Número de ganglios afectados (0, 1-3, ?4).
Brazo A Capecitabina x 8
Brazo B Observación
Seguimiento 5 años
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 9 de 82
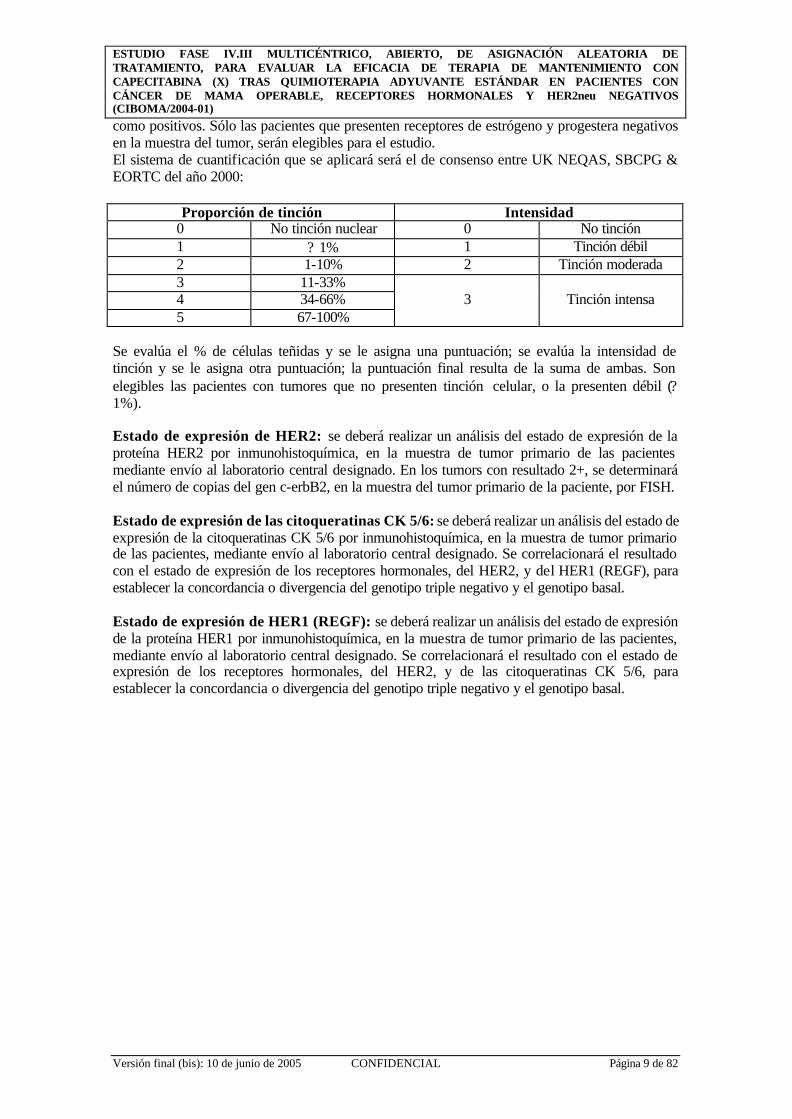
como positivos. Sólo las pacientes que presenten receptores de estrógeno y progestera negativos en la muestra del tumor, serán elegibles para el estudio. El sistema de cuantificación que se aplicará será el de consenso entre UK NEQAS, SBCPG & EORTC del año 2000:
Proporción de tinción Intensidad 0 No tinción nuclear 0 No tinción 1 ? 1% 1 Tinción débil 2 1-10% 2 Tinción moderada 3 11-33% 4 34-66% 5 67-100%
3
Tinción intensa
Se evalúa el % de células teñidas y se le asigna una puntuación; se evalúa la intensidad de tinción y se le asigna otra puntuación; la puntuación final resulta de la suma de ambas. Son elegibles las pacientes con tumores que no presenten tinción celular, o la presenten débil (? 1%). Estado de expresión de HER2: se deberá realizar un análisis del estado de expresión de la proteína HER2 por inmunohistoquímica, en la muestra de tumor primario de las pacientes mediante envío al laboratorio central designado. En los tumors con resultado 2+, se determinará el número de copias del gen c-erbB2, en la muestra del tumor primario de la paciente, por FISH. Estado de expresión de las citoqueratinas CK 5/6: se deberá realizar un análisis del estado de expresión de la citoqueratinas CK 5/6 por inmunohistoquímica, en la muestra de tumor primario de las pacientes, mediante envío al laboratorio central designado. Se correlacionará el resultado con el estado de expresión de los receptores hormonales, del HER2, y del HER1 (REGF), para establecer la concordancia o divergencia del genotipo triple negativo y el genotipo basal. Estado de expresión de HER1 (REGF): se deberá realizar un análisis del estado de expresión de la proteína HER1 por inmunohistoquímica, en la muestra de tumor primario de las pacientes, mediante envío al laboratorio central designado. Se correlacionará el resultado con el estado de expresión de los receptores hormonales, del HER2, y de las citoqueratinas CK 5/6, para establecer la concordancia o divergencia del genotipo triple negativo y el genotipo basal.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 10 de 82
2. INDICE
1.- RESUMEN DEL ESTUDIO..................................................................................... 2
2. INDICE...................................................................................................................... 10
PÁGINA DE FIRMAS DEL PROTOCOLO ............................................................. 14
DECLARACIÓN DEL INVESTIGADOR PRINCIPAL Y EQUIPO..................... 15
1. JUSTIFICACIÓN..................................................................................................... 16
1.1 Antecedentes............................................................................................................ 16
1.2 Quimioterapia adyuvante en el tratamiento del cáncer de mama................................. 17 1.3 Quimioterapia adyuvante en cáncer de mama operable y ganglios positivos o ganglios negativos y alto riesgo ................................................................................................... 18
1.4 Capecitabina........................................................................................................... 20
1.5 Farmacología de capecitabina.................................................................................. 20
1.6 Eficacia antitumoral preclínica de capecitabina. ....................................................... 21 1.7 Estudios clínicos Fase I con capecitabina: esquemas testados, dosis máxima tolerada y dosis recomendada........................................................................................................ 21
1.8 Estudios clínicos Fase I con capecitabina: Farmacocinética de capecitabina en el ser humano......................................................................................................................... 22
1.9 Capecitabina en monoterapia. Estudios Fase II en cáncer de mama............................ 23
1.10 Capecitabina en el tratamiento neoadyuvante del cáncer de mama ........................... 24
1.11 Polimorfismos relacionados con la eficacia y efectividad de la capecitabina ............. 25
1.12 Cáncer de mama de tipo basal................................................................................ 27
1.13 Justificación para el estudio actual......................................................................... 28
2 OBJETIVOS Y PROPÓSITO DEL ESTUDIO...................................................... 29
2.1 Objetivo Primario:.................................................................................................. 29
2.2 Objetivos secundarios:............................................................................................. 29
2.3 Objetivos terciarios: ............................................................................................... 29
3. SELECCIÓN DE LOS PACIENTES ..................................................................... 30
3.1 Criterios de inclusión............................................................................................... 30
3.2 Criterios de exclusión .............................................................................................. 30
4. ESTRATIFICACIÓN Y ASIGNACIÓN ALEATORIA DE BRAZO DE TRATAMIENTO.......................................................................................................... 33
4.1 Calendario de registro y asignación aleatoria en el estudio ........................................ 33
4.2 Procedimiento de asignación aleatoria del tratamiento .............................................. 33

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 11 de 82
4.3 Grupos de tratamiento ............................................................................................. 33
4.4 Estratificación ......................................................................................................... 34
5. TRATAMIENTO DE LOS PACIENTES .............................................................. 35
5.1 Aportación de los fármacos...................................................................................... 35
5.2 Administración de capecitabina ................................................................................ 35 5.3 Medicación concomitante (sólo aplicable a pacientes del brazo de tratamiento con capecitabina)................................................................................................................ 36
Halopurinol.................................................................................................................. 36
Metronidazol................................................................................................................. 36
Cimetidina .................................................................................................................... 36
Factores de crecimiento hematopoyético......................................................................... 36
Laxantes....................................................................................................................... 36
5.4 Ajustes de dosis permitidos....................................................................................... 37
Toxicidad cutánea: síndrome mano-pié grado 2/3 ........................................................... 38 5.4.1 Advertencias y precauciones ......................................................................... 38
5.5 Contabilidad de la medicación.................................................................................. 39
5.6 Quimioterapia adyuvante estándar............................................................................ 39
6. EVALUACIÓN DE LA EFICACIA....................................................................... 40
6.1 Variable principal: evaluación de la eficacia del tratamiento. .................................... 40 6.1.1 Recurrencia objetiva:..................................................................................... 40 6.1.2 Recurrencia local: .......................................................................................... 40 6.1.3 Recurrencia regional: ..................................................................................... 40 6.1.4 Recurrencia a distancia:................................................................................. 40 6.1.5 Segundo tumor primario:............................................................................... 41
6.2 Variable secundaria: Supervivencia Global a los 5 años............................................ 41
6.3 Variable secundaria: evaluación de la seguridad del tratamiento. .............................. 41 6.3.1 Seguridad clínica ........................................................................................... 41
6.4 Variable terciaria (en poblaciones específicas de pacientes): ..................................... 42
6.5 Variable terciaria:................................................................................................... 43
7.1 Calendario de pruebas ............................................................................................. 44
7.2 Criterios de retirada y análisis previstos de retiradas y abandonos............................. 45
7.3 Terapia después de suspender el tratamiento del protocolo ........................................ 45
7.4 Acontecimientos adversos......................................................................................... 45
8. RECOGIDA Y PROCESAMIENTO DE LOS DATOS DEL ESTUDIO ........... 48
8.1 Monitorización, auditoria e inspección...................................................................... 48
8.2 Registro de los datos................................................................................................ 48

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 12 de 82
8.3 Identificación de los datos que se pueden recoger directamente en el CRD y que serán considerados así documentos fuente. .............................................................................. 49
9. CONSIDERACIONES ESTADÍSTICAS............................................................... 50
9.1 Poblaciones a analizar............................................................................................. 50
9.2 Métodos estadísticos................................................................................................ 50
9.3 Evaluación de la seguridad ...................................................................................... 50
9.4 Determinación del tamaño de la muestra................................................................... 51
9.5 Análisis estadístico de los datos de farmacogenética de TS y MTHFR......................... 51
8 ASPECTOS PRÁCTICOS................................................................................... 53
10.1 Comité Independiente de Evaluación de Datos (IDMC)........................................... 53 10.1.1 Composición y misión del IDMC........................................................... 53 10.1.2 Reuniones del IDMC .............................................................................. 53 10.1.3 Recomendaciones del IDMC ....................................................................... 53
10.2 Presupuesto del estudio ......................................................................................... 53
10.3 Seguro de responsabilidad civil .............................................................................. 53
10.4 Archivo de documentación...................................................................................... 54
10.5 Condiciones de publicación .................................................................................... 54
11. ASPECTOS ÉTICOS ............................................................................................. 55
11.1 Declaración de Helsinki..................................................................................... 55
11.3 Confidencialidad............................................................................................... 55
11.5 Modificaciones del protocolo ............................................................................. 55
11.6 Identificación de los pacientes............................................................................ 56
55. BIBLIOGRAFÍA .............................................................................................. 57
APÉNDICE 1. Regímenes mínimos aceptables de quimioterapia para la participación en el estudio CIBOMA 2004-01 ........................................................... 62
APÉNDICE 2: ÍNDICE DEL ESTADO FUNCIONAL DE KARNOFSKY Y CRITERIOS DEL EASTERN COOPERATIVE ONCOLOGY GROUP (ECOG)........................................................................................................................................ 63
APÉNDICE 3: TNM DE MAMA (American Joint Committee on Cancer 2002) .. 64
APÉNDICE 4: MODELO DE CONSENTIMIENTO INFORMADO DE LA PACIENTE.................................................................................................................... 66
APÉNDICE 5: HOJA DE NOTIFICACION DE LOS ACONTECIMIENTOS ADVERSOS................................................................................................................... 73
APÉNDICE 6: LISTA DE PAÍSES PARTICIPANTES ........................................... 75

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 13 de 82
APÉNDICE 7: REGISTRO DE ESTADO MENSTRUAL ...................................... 78
APÉNDICE 8: CRITERIOS DE COCKROFT Y GAULT...................................... 79
APÉNDICE 9: CÁLCULO DE LA DOSIS DE CAPECITABINA EN FUNCIÓN DE LA SUPERFICIE CORPORAL (SÓLO COMPRIMIDOS DE 500 MG)........ 80
APÉNDICE 10: INTERRUPCIÓN Y EXTENSIÓN DEL PERIODO DE REPOSO EN EL TRATAMIENTO CON CAPECITABINA................................................... 82

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 14 de 82
PÁGINA DE FIRMAS DEL PROTOCOLO
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA
DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE,
RECEPTORES HORMONALES Y HER2neu NEGATIVOS CIBOMA/2004-01
Quimioterapia vs. observación Nº EudraCT: 2005-002838-36
Aprobado por: Presidente de la Coalición Iberoamericana de Investigación en Oncología Mamaria (CIBOMA) Prof. Dr. Miguel Martín _____________________________________ ____________ Firma: Fecha: Aprobado por: Coordinador de investigadores en España Dra. Ana Lluch _____________________________________ ____________ Firma: Fecha: Aprobado por: Coordinador de investigadores en América Dra. Laura Torrecillas _____________________________________ ____________ Firma: Fecha: Aprobado por: Coordinador de investigadores en América Dr. Carlos H. Barrios _____________________________________ ____________ Firma: Fecha:

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 15 de 82
DECLARACIÓN DEL INVESTIGADOR PRINCIPAL Y EQUIPO En referencia al protocolo
“ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON
CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS”
CIBOMA/2004-01 Quimioterapia vs. Observación Nº EudraCT: 2005-002838-36
Estoy de acuerdo en que contiene todos los detalles necesarios para realizar este estudio. Llevaré a cabo el estudio según se explica en este documento y en cumplimiento con las Buenas Prácticas Clínicas y de la Declaración de Helsinki (www.wma.net). Procuraré que la inclusión de los pacientes en el estudio finalice en enero de 2008. Facilitaré a todos los médicos responsables ante mí, que participen en el estudio, copias del protocolo y de toda la información sobre el fármaco relativa a la experiencia preclínica y clínica anterior que me ha proporcionado CIBOMA. Comentaré este material con los médicos, para tener la seguridad de que están debidamente informados sobre el fármaco y la realización del estudio. Acepto llevar un registro de toda la información sobre el paciente (cuadernos de recogida de datos y consentimiento informado del paciente), impresos de envío y devolución del fármaco y demás información recopilada durante el estudio, de acuerdo con los requisitos legales. ______________________________________ Investigador (NOMBRE EN LETRAS DE IMPRENTA) ______________________________________ Firma del investigador Fecha ______________________________________ Co-Investigador (NOMBRE EN LETRAS DE IMPRENTA) ______________________________________ Firma del co-investigador Fecha ______________________________________ Co-Investigador (NOMBRE EN LETRAS DE IMPRENTA) ______________________________________ Firma del co-investigador Fecha

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 16 de 82
1. JUSTIFICACIÓN 1.1 Antecedentes El cáncer de mama es el tipo de neoplasia más frecuente entre las mujeres de todo el mundo. En 1.999 se diagnosticaron más de 796.000 nuevos casos y 314.000 fallecimientos debidos a la enfermedad (14,1%). Durante el año 2.000 se registraron en los Estados Unidos 182.800 nuevos casos (30,4% de todas las neoplasias en mujeres estadounidenses) y 40.800 muertes (15,2% de todos los fallecimientos por cáncer). Se calcula que aproximadamente un 30% de las pacientes afectadas en Estados Unidos, fallecerá debido a la enfermedad, y que 1 de cada 8 mujeres desarrollará cáncer de mama a lo largo de su vida. En la Comunidad Europea, se registraron 220.836 nuevos casos en 1.997, lo que suponía 51,67 casos de cáncer de mama por cada 100.000 habitantes, y 74.984 fallecimientos (16,06 muertes por cáncer de mama por cada 100.000 habitantes). Se estima que 1 de cada 10 mujeres europeas desarrollará un cáncer de mama a lo largo de su vida. En España, en 1.997 se registraron 15.906 nuevos casos de cáncer de mama, lo que constituye 69,98 casos de cáncer de mama por cada 100.000 habitantes, con 5.766 fallecimientos (22,67 por cada 100.000 habitantes). Las autoridades calculan una tasa de mortalidad anual por cáncer de mama de 29,3 por 100.000 mujeres. Se estima que 1 de cada 20 mujeres españolas desarrollará cáncer de mama antes de los 75 años. El cáncer de mama es la primera causa de muerte por cáncer entre las mujeres españolas, con una tasa de mortalidad ajustada del 17,4%. En México, se estima que en el año 2010 habrá una tasa de mortalidad de 13 por 100.000 mujeres adultas y cerca de 4.500 defunciones por año por esta causa. En Colombia, el cáncer de mama, representa la tercera causa de muerte por tumores malignos, después del cáncer gástrico y el cáncer de cuello uterino. La incidencia ponderada en las mujeres de 30-50 años es de 80 por cada 100.000 mujeres y el 80% de los casos se encuentran en estado avanzado, con una esperanza de vida limitada. En Cuba, una mujer de cada 9 puede padecer de esta enfermedad, constituyendo el cáncer de mama la primera causa de muerte por cáncer entre mujeres en el país. En Perú se registraron en el año 2000, 7.837 fallecimientos entre mujeres por procesos neoplásicos, con 736 muertes por cáncer de mama. En Venezuela se ha observado un aumento progresivo de su incidencia siendo superado sólo por el cáncer de cuello uterino. La tasa estimada para la incidencia es de 28,08 por cada 100.000 mujeres; la tasa acumulativa se estima en 3,10% lo cual indica que una de cada 33 mujeres en Venezuela tendría cáncer de mama en el curso de su vida. En el año 2000 se produjeron 1.107 defunciones por esta patología, se diagnosticaron 2.704 casos y se ubica en el tercer lugar en mortalidad por cáncer, con 9,22 muertes por 100.000 mujeres. En Argentina, se ha detectado un aumento en la incidencia de mortalidad por cáncer de mama desde el 20,9 por cada 100.000 mujeres, registrado en 1988, al 26,7 por 100.000 mujeres, del periodo 1997-2000. En Costa Rica, la tasa de mortalidad por cáncer de mama registrada en 1998 fue de 9 por 100.000 mujeres. Todos los datos anteriormente expuestos proceden de organismos oficiales. Parece claro que el cáncer de mama se ha convertido en un problema sanitario, social y económico de primer orden en todos los países, o lo será en breve.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 17 de 82
1.2 Quimioterapia adyuvante en el tratamiento del cáncer de mama La cirugía es la principal opción terapéutica en pacientes con cáncer de mama. La cirugía y/o la radioterapia pueden controlar la enfermedad locorregional en la mayoría de los pacientes. No obstante, más del 60% de las afectadas morirán finalmente a causa de la diseminación de la enfermedad1. En los últimos años, se ha producido un incremento en la administración de tratamientos adyuvante hormonales o citostáticos. Los estudios que se llevan a cabo en el momento actual demuestran que el tratamiento adyuvante puede prolongar el tiempo hasta la recurrencia de la enfermedad y probablemente también la supervivencia, en algunos grupos de pacientes2,3. La quimioterapia adyuvante es aquella que se administra después de la cirugía primaria, con el fin de controlar las micro-metástasis clínicamente ocultas. Aunque todavía no se ha identificado el protocolo óptimo, diversos regímenes de quimioterapia han demostrado su eficacia en el tratamiento adyuvante del cáncer de mama. Fundamentalmente se agrupan en regímenes sin antraciclinas, como el CMF (ciclofosfamida, metrotexato, 5-FU), y regímenes que contienen antraciclinas tales como el AC (doxorubicina, ciclofosfamida), CAF (ciclofosfamida, doxorubicina, 5-FU), FAC (5-FU, doxorubicina, ciclofosfamida), AVCF (doxorubicina, vincristina, ciclofosfamida, 5-FU) o FEC (5-FU, epirubicina, ciclofosfamida)4-10. Se han realizado diversos ensayos clínicos aleatorizados para comparar el régimen CMF, o sus variantes, con poliquimioterapias que contienen antraciclinas. La superioridad de la poliquimioterapia con antraciclinas parece ser real, pero modesta6-10. Los resultados del meta-análisis llevado a cabo por el Early Breast Cancer Trialists’ Collaborative Group han confirmado que en general, tanto en las pacientes con ganglios linfáticos axilares negativos como en las que tienen ganglios axilares positivos y criterios de alto riesgo, la quimioterapia adyuvante mejora significativamente la supervivencia libre de enfermedad y la supervivencia global 11. Entre las conclusiones que se extraen de los resultados de estos estudios, se confirma que es mejor administrar 6 ciclos de tratamiento en lugar de 3. Entre los nuevos quimioterápicos que surgieron en la década de los 90, destacan los taxanos como compuestos muy eficaces. Sin embargo, a pesar de que la tendencia de los resultados sugiere que estos agentes podrían suponer un avance significativo en el manejo del cáncer de mama, aún queda por definir el impacto de los mismos en la historia natural de la enfermedad. Se han abordado 2 estrategias diferentes para evaluar el papel potencial de los taxanos en el tratamiento adyuvante, que han quedado reflejadas en varios ensayos clínicos fase III de grandes proporciones: a) La primera estrategia está relacionada con el concepto de quimioterapia secuencial, estrategia que se está investigando tanto con paclitaxel como con docetaxel. Algunos de los grupos cooperativos que han realizado ensayos clínicos siguiendo esta estrategia son el CALGB (Cancer Leukemia Group B) con el esquema ATC (doxorubicina seguido por paclitaxel seguido por ciclofosfamida), el MD Anderson con TFAC (paclitaxel seguido de FAC), el CALGB y NSABP (National Surgical Adjuvant Breast and Bowel Project) con AC seguido de paclitaxel o docetaxel, el Breast Adjuvant Study Team y el IBCSG (International Breast Cáncer Study Group) con AT (docetaxel) seguido de CMF, el French Cooperative Group con el esquema FEC(5-fluorouracilo, epidoxorubicina, ciclofosfamida) seguido por docetaxel, el BCIRG (Breast Cancer International Research Group) con AC seguido de docetaxel + trastuzumab (en pacientes HER2neu positivo) y GEICAM con FEC seguido de paclitaxel semanal12.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 18 de 82
b) La segunda estrategia se basa en el concepto clásico de la poliquimioterapia, en la que se estudian casi exclusivamente combinaciones basadas en el tratamiento con docetaxel. Se han desarrollado protocolos comparativos de TAC (docetaxel, doxorubicina, ciclofosfamida) frente a FAC (BCIRG-001)15, llevado a cabo por el BCIRG en pacientes con ganglios linfáticos axilares positivos, o el que compara el régimen AT (docetaxel) con AC coordinado por el ECOG (Eastern Cancer Oncology Group) en pacientes con 1 a 3 ganglios positivos o con ganglios negativos y criterios de alto riego. Asimismo, se están realizando ensayos para estudiar si la triple combinación es mejor que la quimioterapia secuencial, como el trabajo coordinado por el BCIRG (BCIRG-005) en el que se enfrenta TAC a AC seguido de T en pacientes con ganglios positivos y HER2neu negativo. 1.3 Quimioterapia adyuvante en cáncer de mama operable y ganglios positivos o ganglios negativos y alto riesgo Los resultados más recientes de dos de los regímenes de combinaciones con taxanos han demostrado una mayor eficacia en la población de pacientes con cáncer de mama operable. Se trata del esquema de dosis densas bisemanal de AC seguido de paclitaxel, basado en los resultados del estudio GALGB-9741, y del esquema TAC (docetaxel, doxorubicina, ciclofosfamida), basado en los resultados del estudio BCIRG001. La comparación indirecta de los resultados de ambos estudios sugiere una eficacia y tolerabilidad similares, y ambos muestran un incremento en la supervivencia global, frente a esquemas estándar. En ambos casos se trata de estudios multicéntricos de gran tamaño de muestra, por lo que los resultados se pueden considerar evidencia científica de tipo 1. Estudio CALGB -974114: Se incluyeron 2.005 pacientes con ganglios positivos con el objetivo de determinar si la densidad de dosis de los fármacos mejoraría la supervivencia libre de enfermedad y la supervivencia global. Fueron asignados de forma aleatoria a 4 brazos, 2 secuenciales y 2 concurrentes: Ax4 ? T (paclitaxel) x4 ? Cx4 (una rama administrando los ciclos cada 2 semanas y otra cada 3 semanas) y ACx4 ? Tx4 (una rama administrando los ciclos cada 2 semanas y otra cada 3 semanas). Los resultados a los 36 meses de seguimiento del estudio CALGB-9741 mostraban un incremento en la supervivencia libre de enfermedad y en la supervivencia global del esquema de dosis densas (A->C->P bisemanal) del 85% vs. 81% (p=0,01), y del 92% vs. 90% (p=0,013), respectivamente, frente al esquema de tratamiento convencional (administración cada 21 días). Estudio BCIRG 00115: En el estudio BCIRG 001 se comparaban 6 ciclos de TAC (docetaxel) vs. 6 de FAC, en 1.491 pacientes con ganglios positivos. Los resultados a los 55 meses de seguimiento del estudio BCIRG-001 confirman la superioridad del esquema TAC en la supervivencia global, frente a FAC (razón de riesgos de 0,7 [0,53-0,91], p=0,0080). Con respecto a la supervivencia libre de enfermedad, se ha realizado el análisis por subgrupos (número de ganglios afectos, estado de los receptores hormonales), con los siguientes resultados: N= 1.491 Razón de riesgos (Hazart Ratio) Valor p SLE Todos los pacientes 0,72 (0,59-0,88) 0,0010 1-3 ganglios (n=923) 0,61 (0,46-0,82) 0,0009 ? 4 ganglios (n=568) 0,82 (0,63-1,08) 0,1629 RH positivos 0,73 (0,57-0,94) 0,0132 RH negativos 0,66 (0,47-0,93) 0,0163

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 19 de 82
Como se observa, los pacientes que más se benefician del tratamiento con TAC son los pacientes con menor número de ganglios afectados, y los pacientes con receptores hormonales negativos. Los resultados de los estudios anteriormente descritos han servido de base a una nueva generación de ensayos clínicos en fase III, de asignación aleatoria de tratamiento, que se encuentran actualmente en marcha, y que se resumen a continuación: Estudio US Oncology 01-062 Este estudio reclutará 1.810 pacientes con cáncer de mama operable y ganglios positivos o con ganglios negativos y criterios de alto riesgo. Las pacientes elegibles se randomizan para recibir 4 ciclos de AC seguido de 4 ciclos de docetaxel (ACx4->T4), frente a 4 ciclos de AC seguidos de 4 ciclos de docetaxel y capecitabina (ACx4->TXx4). Estudio SWOG-S0221 En este estudio se reclutarán 4.500 pacientes con cáncer de mama operable y ganglios positivos o con ganglios negativos y criterios de alto riesgo. Las pacientes elegibles se randomizan para recibir uno de los 4 brazos de tratamientos siguientes:
- 6 ciclos de AC bisemanal seguido de 6 ciclos de paclitaxel bisemanal (ACx6->Px6) - 15 ciclos de AC semanal (con ciclofosfamida oral días 1-7) seguido de 6 ciclos de
paclitaxel bisemanal (ACx15->Px6) - 6 ciclos de AC bisemanal seguido de 12 ciclos de paclitaxel semanal (ACx6->Px12) - 15 ciclos de AC semanal (con ciclofosfamida oral días 1-7) seguido de 12 ciclos de
paclitaxel semanal (ACx15->Px12) Estudio NSABP B-38 En este estudio se reclutarán 4.500 pacientes con cáncer de mama operable y ganglios positivos. Las pacientes elegibles se randomizan para recibir uno de los 3 brazos de tratamientos siguientes:
- 6 ciclos de TAC (docetaxel, doxorubicina, ciclosfosfamida) cada 21 días (TACx6). - 4 ciclos de AC bisemanal seguido de 4 ciclos de paclitaxel bisemanal (ACx4->Px4) - 4 ciclos de AC bisemanal seguido de 4 ciclos de paclitaxel/gemcitabina bisemanal
(ACx4->PGx4). Estudio MA.21 del NCIC CTG En este estudio se reclutarán 1.500 pacientes con cáncer de mama operable y ganglios positivos o con ganglios negativos y criterios de alto riesgo. Las pacientes elegibles se randomizan para recibir uno de los 3 brazos de tratamientos siguientes:
- 6 ciclos de epirubicina y fluorouracilo (días 1-8) con ciclofosfamida oral (días 1-14) cada 28 días (FECx6).
- 6 ciclos de EC bisemanal seguido de 4 ciclos de paclitaxel cada 3 semanas (ECx6->Px4).
- 4 ciclos de AC cada 3 semanas seguido de 4 ciclos de paclitaxel cada 3 semanas (ACx4->Px4).
CALGB 49907: Se trata de un ensayo fase III que incluirá pacientes > 65 años con cáncer de mama operable y ganglios linfáticos positivos o negativos con factores de alto riesgo. Se propone dar una alternativa viable a ese gran grupo de pacientes mayores que con frecuencia reciben tratamientos subóptimos por temor a un incremento de toxicidad423. Las pacientes serán randomizadas a unos de los siguientes brazos: -CMF oral x 6 ciclos

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 20 de 82
-AC x 4 ciclos -Capecitabina x 6 ciclos. Estudio GEICAM/2003-10 En este estudio se reclutarán 1.302 pacientes con cáncer de mama operable y ganglios positivos. Las pacientes elegibles se randomizan para recibir uno de los 2 brazos de tratamientos siguientes:
- 4 ciclos de epirubicina y ciclofosfamida cada 21 días seguido de 4 ciclos de docetaxel cada 21 días (ECx4->Tx4).
- 4 ciclos de epirubicina y docetaxel cada 21 días seguido de 4 ciclos de capecitabina días 1-14 de cada ciclo de 21 días (ETx4 -> Xx4).
Todos estos estudios tienen en común una serie de criterios:
- Los esquemas de combinaciones de antraciclinas y ciclofosfamida se administran bien en dosis densas, o como esquema metronómico.
- Se administran un mínimo de 4 ciclos de quimioterapia (doxorubicina + docetaxel), y se tiende a alargar los periodos de tratamiento, incrementando el número de ciclos administrados.
- Se añaden a los fármacos ya conocidos en el tratamiento adyuvante (antraciclinas y taxanos) los nuevos productos que ya han demostrado eficacia en el tratamiento de cáncer de mama avanzado o metastásico (capecitabina, gemcitabina).
1.4 Capecitabina. La capecitabina es un carbamato de fluoropirimidina oral que se activa preferentemente en el tumor a través de una cascada de 3 enzimas, que proporcionan así niveles altos y prolongados de la fracción activa, 5-fluoruracilo (5-FU) en las células tumorales. El fluoruracilo es uno de los agentes citotóxicos más extensamente usados, pero su eficacia está limitada por su falta de selectividad. La capecitabina se desarrolló mediante investigación como un agente selectivo del tumor que alteraría favorablemente la relación riesgo-beneficio, especialmente en el contexto de la quimioterapia en cáncer de mama avanzado y resistente31. 1.5 Farmacología de capecitabina. Después de la administración oral, la capecitabina se absorbe sin cambios en la vía gastrointestinal y se metaboliza en el hígado por 60kDa carboxilesterasa, a 5´-DFCR. Después se convierte en 5´-DFUR por la citidina deaminasa localizada en el hígado y en los tejidos tumorales. Ya en el tumor se produce el metabolismo posterior de 5´-DFUR a 5-FU bajo la acción de la timidina fosforilasa (dThdPasa). Por lo tanto, se minimiza la exposición de los tejidos sanos a 5-FU sistémico16. Se ha sabido recientemente que la enzima responsable de la conversión catalítica final de capecitabina a 5-FU (dThdPasa) es de estructura y función idéntica al factor angiogénico asociado a tumores, el factor de crecimiento celular endotelial derivado de las plaquetas (PD-ECGF)17-19. PD-ECGF es un polipéptido 55 KDa que existe in vivo como un homodímero. Se aisló originalmente a partir de las plaquetas como un mitógeno endotelial y factor angiogénico20,21.PD-ECGF ha demostrado ser un importante factor angiogénico en cáncer de mama. Su expresión se correlaciona con la intensidad de la angiogénesis, con un aumento del tamaño del tumor y de la capacidad invasiva de éste, así como con un peor pronóstico21,22. Las áreas del tumor con inferior perfusión, definida por hipoxia y acidosis, tienen una expresión muy aumentada de PD-ECGF20. El hecho de que a nivel tumoral y, en especial en tumores más agresivos, existan niveles elevados de PD-ECGF/dThdPasa, responsables de la activación última de la capecitabina, apoyan la eficacia antitumoral de éste fármaco.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 21 de 82
1.6 Eficacia antitumoral preclínica de capecitabina. Capecitabina demostró citotoxicidad únicamente después de su conversión a 5´-DFUR y 5-FU. En 12 modelos de xenoinjertos de cáncer humano probados en ratones, capecitabina demostró ser siempre más activo que 5-FU. Además, mostró actividad en xenoinjertos resistentes a 5-FU. Los índices terapéuticos fueron mucho más favorables que los observados con 5-FU. La eficacia antitumoral en ratones se correlacionó con los niveles de 5-FU en el tumor y 5´-DFUR en sangre. Además, también en ratones, capecitabina demostró actividad anticaquética, normalizando la pérdida de peso, la pérdida de tejido adiposo y los episodios de hipoglucemia. En este mismo modelo, capecitabina demostró acción antimetastática en dosis 40 a 50 veces inferiores a las requeridas para la acción contra la enfermedad primaria 16,23. Los estudios preclínicos han revelado que la actividad antitumoral de capecitabina depende de la dosis total administrada, en tanto que la pauta (administración diaria ininterrumpida; días 1-5 cada semana; días 1-14 cada 3 semanas) no influye en su eficacia antitumoral24. 1.7 Estudios clínicos Fase I con capecitabina: esquemas testados, dosis máxima tolerada y dosis recomendada La dosis máxima tolerada (DMT) de capecitabina en monoterapia continua o intermitente25-27 o en combinación con leucovorin28 se definió en cuatro estudios Fase I (SO14693, SO14794, JO14865, SO14798). Las dosis recomendadas determinadas por dichos estudios se han utilizado posteriormente con resultados satisfactorios en estudios fases II y III. En general, las tres pautas utilizadas (régimen continuo, intermitente o asociado a leucovorín) demostraron eficacia antitumoral significativa, incluyendo respuestas completas. Las tres pautas probaron ser tolerables con escasos acontecimientos grado 3/4 que fueron manejables con modificación de la dosis y/o intervención sintomática. La valoración global llevada a cabo por los investigadores y el promotor sobre la base de dichos estudios fase I y a un estudio fase II de distribución aleatoria en cáncer colorrectal29 fue que la pauta intermitente era la más prometedora, al asociar un perfil de toxicidad razonable y un mayor tiempo a progresión de la enfermedad, al tiempo que permitía administrar una mayor intensidad de dosis al paciente (aproximadamente 25% superior a la de la pauta continua). La ventaja adicional de los períodos “sin fármaco” con esta pauta también se consideró como más satisfactoria por los pacientes. Del estudio fase I europeo con esquema intermitente (2 semanas de tratamiento y una de descanso) se definió una DMT de 1500 mg/m2 dos veces al día, mientras que la dosis recomendada para estudios fase II se estableció en 1250 mg/m2 dos veces al día 26. En este mismo trabajo, en el que participaron siete pacientes con cáncer de mama y 17 pacientes con cáncer colorrectal altamente pretratados, se describieron cuatro respuestas objetivas (1RC y 3 RP). Los tumores que respondieron fueron cánceres de mama, esófago, colon y recto. Un estudio en Fase I de tratamiento de combinación de capecitabina con paclitaxel30 determinó una dosis recomendada para estudios fase II de 665 mg/m2/2 veces al día de capecitabina oral, dividida en 2 tomas diarias y de forma continua, y de 175 mg/m2 de paclitaxel en una infusión de 3 horas e.v. cada 3 semanas. No se objetivaron interacciones farmacocinéticas clínicamente relevantes entre ambos fármacos. No se observaron respuestas (15 de 17 pacientes habían sido claramente refractarios a 5-FU y 13 de 17 pacientes tenían neoplasias consideradas intrínsicamente resistentes a taxanos). El estudio SO15304 en Fase I investigó la combinación de capecitabina administrado los días 1 a 14 junto con docetaxel administrado el día 1 de cada ciclo cada tres semanas31. El incremento progresivo de dosis se produjo en dos fases. Inicialmente, la dosis de docetaxel se aumentó (75, 85 y 100 mg/m2) en combinación con una dosis fija de capecitabina (825 mg/m2/2 veces al día). Posteriormente, se aumentó la dosis de capecitabina (1.000 y 1.200 mg/m2/2 veces al día) en combinación con una dosis fija de docetaxel definida como tolerable en la primera fase. Se incluyeron 33 pacientes (15 hombres, 18 mujeres) distribuidos en siete niveles de dosificación: 75 (4 pacientes), 85 (6 pacientes) y 100 (6 pacientes) mg/m2 de docetaxel en combinación con 825 mg/m2/2 veces al día de capecitabina, 100 (5 pacientes) mg/m2 de docetaxel en combinación con 1.000 mg/m2/2 veces al día de capecitabina, y después 1.000 mg/m2/2 veces al

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 22 de 82
día (6 pacientes) y 1.250 mg/m2/2 veces al día (6 pacientes) capecitabina en combinación con 75 mg/m2 de docetaxel. Se observó neutropenia grado 4 no complicada (afebril y de duración inferior a 7 días) en todos los niveles de dosificación. Se observó un caso de neutropenia grado 3-4 asociada a fiebre grado 2 en los últimos ciclos de tratamiento de cada uno de los niveles de dosificación 85/820, 100/825 y 100/1.000 mg/m2 docetaxel/capecitabina. Un paciente con los niveles de dosificación 100/1.000 y 85/825 mg/m2 docetaxel/capecitabina experimentó mucositis grado 3 NCIC/ CTC. Los 5 pacientes tratados en el nivel de dosificación de 100/1.000 mg/m2 docetaxel/capecitabina informaron de astenia crónica grado 2 y cumplieron la definición de DTM. Otras toxicidades frecuentes (grado 2) incluyeron alopecia, diarrea, mucositis, síndrome mano-pie/toxicidad en la piel, distrofia de las uñas, náusea y vómito. Requirieron reducción de dosis 7 pacientes, 5 debido a síndrome mano-pie o distrofia en las uñas, uno debido a estomatitis y uno debido a trombocitopenia. Se observaron respuestas parciales en tres de los cuatro pacientes con cáncer de mama y respuestas menores en carcinoma adenoquístico submandibular, carcinoma de pulmón no microcítico, carcinoma colorrectal y un adenocarcinoma de origen primario desconocido. Se observó toxicidad mínima diferente a la neutropenia esperada en el nivel de dosificación de 75/1250 mg/m2 docetaxel/capecitabina, experimentando sólo 2 pacientes astenia grado 2 de corta duración (produciéndose en la interrupción de la comedicación con dexametasona). Por lo tanto, este nivel de dosificación se seleccionó para estudios posteriores. 1.8 Estudios clínicos Fase I con capecitabina: Farmacocinética de capecitabina en el ser humano A partir de los estudios fase I con capecitabina se han obtenido parámetros de farmacocinética del producto. La comunicación de Twelves32 resume la mayoría de datos conocidos hasta el momento. Tales datos se basaron en 32 pacientes que recibieron dosis de 251 a 1255 mg/m2/2 veces al día en dos tomas separadas. La absorción de capecitabina fue rápida y prácticamente completa. Las concentraciones plasmáticas del fármaco en el pico y las de sus dos metabolitos principales (5´-DFCR y 5´-DFUR) se alcanzaron rápidamente (0,5 a 1,5 horas) después de la administración. Menos del 60% de capecitabina y de sus metabolitos se unieron a proteínas plasmáticas. Las concentraciones disminuyeron después exponencialmente con una vida media de 0,5 a 1 hora. Más del 70% de la dosis administrada de capecitabina fue recuperada en la orina. Después de una administración de 829 mg/m2, la mayor AUC se obtuvo para 5´-DFUR (11,8 ?g.mL-1.h, CV= 44%, n=15). Se utilizaron técnicas de regresión logística para evaluar las relaciones entre toxicidad y exposición sistémica. Los dos pacientes con la mayor exposición a 5´-DFUR tuvieron toxicidad Grado 3. Se obtuvieron parámetros farmacocinéticos similares en los días 1 y 15?1. Las características farmacocinéticas de capecitabina y sus metabolitos fueron proporcionales a la dosis hasta 1.657 mg/m2/día. Un estudio ha correlacionado la presencia de alimento con una modificación de varios parámetros farmacocinéticos (descenso de la concentración plasmática máxima y de la AUC e incremento del tiempo a la presentación de la concentración plasmática máxima) con respecto a la administración de capecitabina en ayunas33. Hasta ahora, el resto de estudios clínicos se han realizado en presencia de alimento. Por ello, ante la ausencia de datos de seguridad/eficacia de capecitabina administrada en condiciones de ayuno, la recomendación actual es la de continuar administrando el fármaco 30 minutos después de una ingesta (desayuno o comida). Con el fin de investigar las concentraciones diferenciales de 5-FU (en tumor, tejido sano y plasma) tras la administración de capecitabina, se llevó a cabo un estudio farmacocinético en 19 pacientes con cáncer colorrectal sometidos a cirugía. Después de la administración oral de capecitabina (1.255 mg/m2 dos veces al día durante 5 a 7 días antes de la cirugía), las concentraciones de 5-FU fueron significativamente superiores (relación geométrica media 2,5; CI: 1,5 a 4,1) en el tumor primario que en el tejido normal adyacente34. El hallazgo es consistente con otros estudios. Así, los tumores humanos, en particular el de mama, gástrico, colorrectal, cervical y de ovario han demostrado tener niveles mucho más altos de dThdPasa (responsable de la conversión de 5’-DFUR a 5-FU) que los tejidos normales correspondientes16.
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 23 de 82
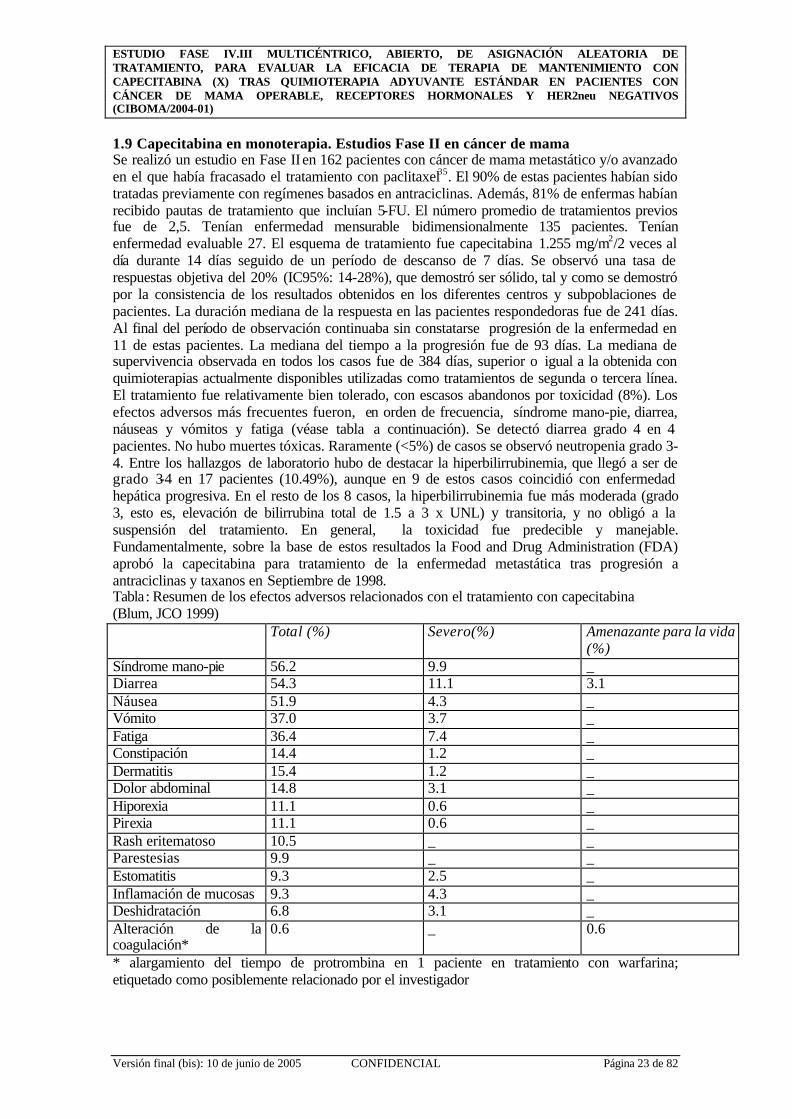
1.9 Capecitabina en monoterapia. Estudios Fase II en cáncer de mama Se realizó un estudio en Fase II en 162 pacientes con cáncer de mama metastático y/o avanzado en el que había fracasado el tratamiento con paclitaxel35. El 90% de estas pacientes habían sido tratadas previamente con regímenes basados en antraciclinas. Además, 81% de enfermas habían recibido pautas de tratamiento que incluían 5-FU. El número promedio de tratamientos previos fue de 2,5. Tenían enfermedad mensurable bidimensionalmente 135 pacientes. Tenían enfermedad evaluable 27. El esquema de tratamiento fue capecitabina 1.255 mg/m2/2 veces al día durante 14 días seguido de un período de descanso de 7 días. Se observó una tasa de respuestas objetiva del 20% (IC95%: 14-28%), que demostró ser sólido, tal y como se demostró por la consistencia de los resultados obtenidos en los diferentes centros y subpoblaciones de pacientes. La duración mediana de la respuesta en las pacientes respondedoras fue de 241 días. Al final del período de observación continuaba sin constatarse progresión de la enfermedad en 11 de estas pacientes. La mediana del tiempo a la progresión fue de 93 días. La mediana de supervivencia observada en todos los casos fue de 384 días, superior o igual a la obtenida con quimioterapias actualmente disponibles utilizadas como tratamientos de segunda o tercera línea. El tratamiento fue relativamente bien tolerado, con escasos abandonos por toxicidad (8%). Los efectos adversos más frecuentes fueron, en orden de frecuencia, síndrome mano-pie, diarrea, náuseas y vómitos y fatiga (véase tabla a continuación). Se detectó diarrea grado 4 en 4 pacientes. No hubo muertes tóxicas. Raramente (<5%) de casos se observó neutropenia grado 3-4. Entre los hallazgos de laboratorio hubo de destacar la hiperbilirrubinemia, que llegó a ser de grado 3-4 en 17 pacientes (10.49%), aunque en 9 de estos casos coincidió con enfermedad hepática progresiva. En el resto de los 8 casos, la hiperbilirrubinemia fue más moderada (grado 3, esto es, elevación de bilirrubina total de 1.5 a 3 x UNL) y transitoria, y no obligó a la suspensión del tratamiento. En general, la toxicidad fue predecible y manejable. Fundamentalmente, sobre la base de estos resultados la Food and Drug Administration (FDA) aprobó la capecitabina para tratamiento de la enfermedad metastática tras progresión a antraciclinas y taxanos en Septiembre de 1998. Tabla: Resumen de los efectos adversos relacionados con el tratamiento con capecitabina (Blum, JCO 1999) Total (%) Severo(%) Amenazante para la vida
(%) Síndrome mano-pie 56.2 9.9 _ Diarrea 54.3 11.1 3.1 Náusea 51.9 4.3 _ Vómito 37.0 3.7 _ Fatiga 36.4 7.4 _ Constipación 14.4 1.2 _ Dermatitis 15.4 1.2 _ Dolor abdominal 14.8 3.1 _ Hiporexia 11.1 0.6 _ Pirexia 11.1 0.6 _ Rash eritematoso 10.5 _ _ Parestesias 9.9 _ _ Estomatitis 9.3 2.5 _ Inflamación de mucosas 9.3 4.3 _ Deshidratación 6.8 3.1 _ Alteración de la coagulación*
0.6 _ 0.6
* alargamiento del tiempo de protrombina en 1 paciente en tratamiento con warfarina; etiquetado como posiblemente relacionado por el investigador

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 24 de 82
Otro estudio fase II se llevó a cabo también en pacientes extensamente pretratadas36(el 100% habían recibido paclitaxel y/o docetaxel , el 92% habían recibido antraciclinas) utilizando una pauta muy similar de capecitabina (1250 mg/m2 2 veces al día, días 1 a 14, cada 3 semanas). Ciento cinco pacientes fueron evaluables para toxicidad, mientras que 100 lo fueron para respuesta. Se objetivó una tasa de respuestas del 18% (16%RP, 2%RC). La toxicidad fue en general moderada, de grado 1-2, y consistió en síndrome mano-pie (42% de pacientes), náuseas/vómitos (40%), diarrea (26%), estomatitis (16%) y letargia (15%). Se registró síndrome mano-pie grado 3 en 13% de las enfermas. Se objetivó, además, emesis y diarrea grado 3 en 5% y 3% de casos, respectivamente. Jakob y col 37 trataron 13 pacientes con el mismo esquema de capecitabina. Todas las pacientes habían recibido antraciclinas, taxanos y quimioterapia a dosis altas. Se obtuvo una tasa de respuestas del 54% (IC 95%:26-81%). La mediana del tiempo a la progresión fue de 128 días. El espectro de toxicidad fue similar al descrito en los estudios previos. Cinco pacientes (37.5%) desarrollaron síndrome mano-pie grado 3. En otro estudio realizado en pacientes pretratadas con antraciclinas, se evaluó paclitaxel a dosis habituales (175 mg/m2 cada 3 semanas) versus capecitabina en pauta intermitente (2510 mg/m2/día en 2 dosis días 1 a 14 cada 3 semanas)38. El estudio se cerró prematuramente por falta de reclutamiento (rechazo de la randomización). Finalmente sólo 22 pacientes recibieron capecitabina y 20 paclitaxel. Se observaron 8 respuestas (36%, IC 17-59%), incluyendo 3 respuestas completas, en el grupo tratado con capecitabina. En el grupo que recibió paclitaxel se objetivaron 4 respuestas parciales (21%, IC 95% 6-46%). Las medianas de los tiempos a la progresión fueron de 92 y 95 días, para capecitabina y paclitaxel, respectivamente. La proporción de pacientes que presentaron toxicidad grado 3-4 fue del 22% en el grupo que recibió capecitabina y del 58% en el grupo tratado con paclitaxel. El estudio, aunque notablemente limitado por el pequeño tamaño muestral, apoya una similar eficacia entre ambos fármacos, con un mejor perfil de tolerabilidad para capecitabina. Consistentemente, en un estudio fase II japonés realizado en 50 pacientes pretratadas de forma heterogénea (exposición previa a antraciclinas 36%, a alquilantes:54%,a 5FU 64%, a hormonas:74%), se describió una tasa de respuestas del 28.3% para el grupo total y del 23.5% para el grupo pretratado con antraciclinas39. Un estudio de distribución aleatoria de tratamiento, efectuado en 95 pacientes no pretratadas40 comparó capecitabina, administrada según el esquema intermitente, con el régimen CMF e.v., administrado éste día 1 cada 3 semanas. La tasa de respuestas con capecitabina y CMF fue del 25 (IC 95%: 14-37%) y del 16% (IC 95%: 5-33%), respectivamente. La mediana del tiempo a la progresión fue superior con capecitabina (132 vs. 94 días). Se objetivó una mayor proporción de toxicidad grado 3 en el grupo tratado con capecitabina (44 vs. 20%). Estas diferencias fueron debidas fundamentalmente a una mayor frecuencia de síndrome mano-pie (16 vs. 0%) y de diarrea (8 vs. 3%) en el grupo que recibió el esquema oral. No se efectuó un análisis estadístico para ninguno de los parámetros de eficacia o toxicidad. Los autores concluyeron que capecitabina mostraba al menos similar eficacia que CMF cuando se administraba como tratamiento de primera línea. Estudios posteriores han confirmado la eficacia y tolerabilidad del tratamiento con capecitabina en cáncer de mama metastásico43. Capecitabina en combinación con docetaxel también ha demostrado ser más eficaz en tratamiento de cáncer de mama metastásico que docetaxel en monoterapia, en términos de supervivencia, y tasas de respuesta44. Dado el papel de capecitabina en cáncer de mama metastásico, como agente altamente eficaz y sin resistencias cruzadas, así como la ventaja de su administración oral, parece justificado el estudio de su administración en el entorno adyuvante. 1.10 Capecitabina en el tratamiento neoadyuvante del cáncer de mama El tratamiento primario del cáncer de mama en estadio II/III, con una combinación de docetaxel y capecitabina rinde tasas de respuestas patológicas y clínicas superiores a otros esquemas, como doxorubicina/ciclofosfamida (AC). En el estudio llevado a cabo por Hong Gi Lee et al45
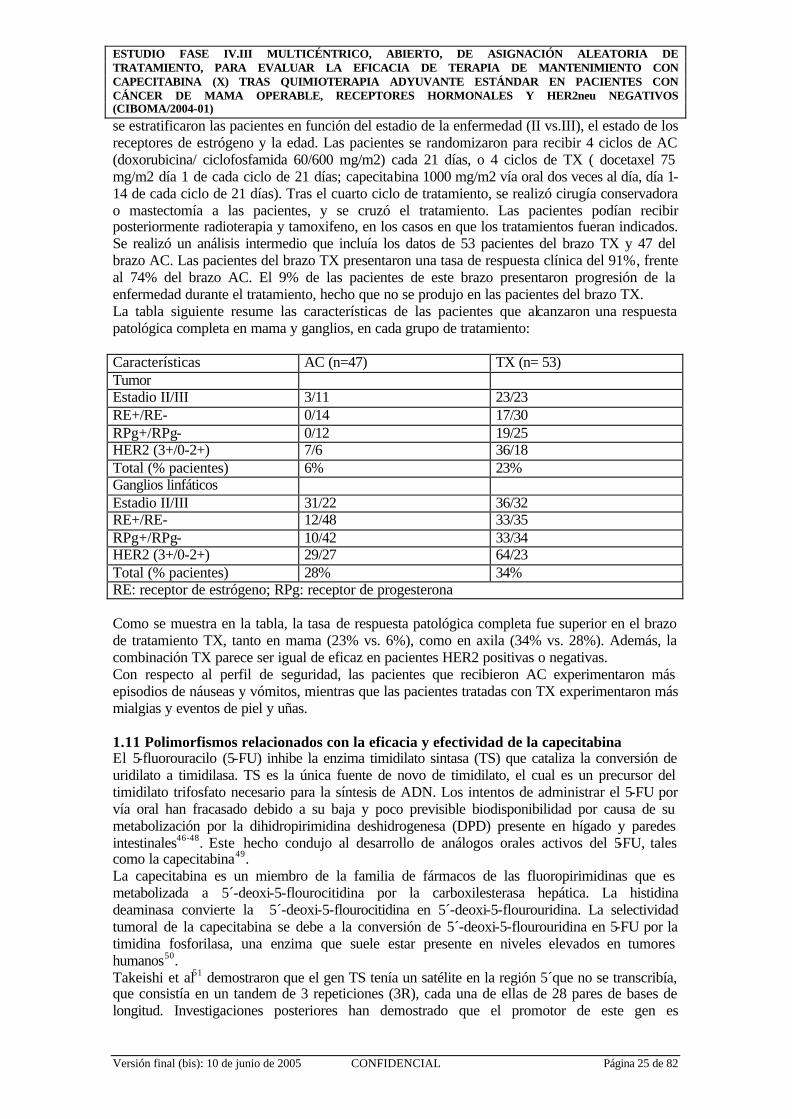
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 25 de 82
se estratificaron las pacientes en función del estadio de la enfermedad (II vs.III), el estado de los receptores de estrógeno y la edad. Las pacientes se randomizaron para recibir 4 ciclos de AC (doxorubicina/ ciclofosfamida 60/600 mg/m2) cada 21 días, o 4 ciclos de TX ( docetaxel 75 mg/m2 día 1 de cada ciclo de 21 días; capecitabina 1000 mg/m2 vía oral dos veces al día, día 1-14 de cada ciclo de 21 días). Tras el cuarto ciclo de tratamiento, se realizó cirugía conservadora o mastectomía a las pacientes, y se cruzó el tratamiento. Las pacientes podían recibir posteriormente radioterapia y tamoxifeno, en los casos en que los tratamientos fueran indicados. Se realizó un análisis intermedio que incluía los datos de 53 pacientes del brazo TX y 47 del brazo AC. Las pacientes del brazo TX presentaron una tasa de respuesta clínica del 91%, frente al 74% del brazo AC. El 9% de las pacientes de este brazo presentaron progresión de la enfermedad durante el tratamiento, hecho que no se produjo en las pacientes del brazo TX. La tabla siguiente resume las características de las pacientes que alcanzaron una respuesta patológica completa en mama y ganglios, en cada grupo de tratamiento: Características AC (n=47) TX (n= 53) Tumor Estadio II/III 3/11 23/23 RE+/RE- 0/14 17/30 RPg+/RPg- 0/12 19/25 HER2 (3+/0-2+) 7/6 36/18 Total (% pacientes) 6% 23% Ganglios linfáticos Estadio II/III 31/22 36/32 RE+/RE- 12/48 33/35 RPg+/RPg- 10/42 33/34 HER2 (3+/0-2+) 29/27 64/23 Total (% pacientes) 28% 34% RE: receptor de estrógeno; RPg: receptor de progesterona Como se muestra en la tabla, la tasa de respuesta patológica completa fue superior en el brazo de tratamiento TX, tanto en mama (23% vs. 6%), como en axila (34% vs. 28%). Además, la combinación TX parece ser igual de eficaz en pacientes HER2 positivas o negativas. Con respecto al perfil de seguridad, las pacientes que recibieron AC experimentaron más episodios de náuseas y vómitos, mientras que las pacientes tratadas con TX experimentaron más mialgias y eventos de piel y uñas.
1.11 Polimorfismos relacionados con la eficacia y efectividad de la capecitabina El 5-fluorouracilo (5-FU) inhibe la enzima timidilato sintasa (TS) que cataliza la conversión de uridilato a timidilasa. TS es la única fuente de novo de timidilato, el cual es un precursor del timidilato trifosfato necesario para la síntesis de ADN. Los intentos de administrar el 5-FU por vía oral han fracasado debido a su baja y poco previsible biodisponibilidad por causa de su metabolización por la dihidropirimidina deshidrogenesa (DPD) presente en hígado y paredes intestinales46-48. Este hecho condujo al desarrollo de análogos orales activos del 5-FU, tales como la capecitabina49. La capecitabina es un miembro de la familia de fármacos de las fluoropirimidinas que es metabolizada a 5´-deoxi-5-flourocitidina por la carboxilesterasa hepática. La histidina deaminasa convierte la 5´-deoxi-5-flourocitidina en 5´-deoxi-5-flourouridina. La selectividad tumoral de la capecitabina se debe a la conversión de 5´-deoxi-5-flourouridina en 5-FU por la timidina fosforilasa, una enzima que suele estar presente en niveles elevados en tumores humanos50. Takeishi et al51 demostraron que el gen TS tenía un satélite en la región 5´que no se transcribía, que consistía en un tandem de 3 repeticiones (3R), cada una de ellas de 28 pares de bases de longitud. Investigaciones posteriores han demostrado que el promotor de este gen es

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 26 de 82
polimórfico, con individuos que tienen sólo 2 repeticiones (2R) y otros que tienen hasta 9 repeticiones (9R)52-54. El número de repeticiones en tandem afecta a la transcripción del gen TS55. Los pacientes homocigóticos para tres repeticiones (3R/3R) tienen niveles mayores de proteína TS que los heterocigóticos 2R/3R. Usando la expresión in vitro de genes con 2R y 3R, se obtuvieron mayores niveles de proteína TS debidos a una mayor eficacia transcripcional del ARN mensajero del 3R, frente a un aumento de producción de ARN mensajero. Diversos estudios han sugerido que los genotipos 3R/3R de la TS en cáncer colorrectal se asocian con una peor respuesta a la quimioterapia con 5-FU. Se sospecha que el motivo de ello es que los genotipos 3R/3R se asocian con niveles más elevados de proteína TS, lo que confiere resistencia al 5FU. En un análisis retrospectivo, en el que se trataron a pacientes con cáncer colorrectal con capecitabina, las tasas de respuesta fueron del 14% en los sujetos con genotipo 3R/3R, comparada con una tasa de respuesta del 80% en sujetos con genotipo 2R/2R56. Estudios recientes han demostrado que los polimorfismos de repetición en tandem de TS contienen posibles sitios de unión denominados E-Box (CACTTG) que unen factores de trascripción por encima del sitio de unión del factor estimulatorio 1 (USF-1) y el factor estimulatorio 2 (USF-2)57. En los genotipos 3R/3R, existen dos sitios de unión E-Box en la segunda y tercera repeticiones, mientras que el genotipo 2R tiene un solo sitio de unión E-Box. La repetición final de ambos genotipos 2R y 3R es imperfecta en el nucleótido 12, que mientras en la primera repetición es una G, en ésta ha sido reemplazado por una C. Se descubrió un polimorfismo en la posición 12 G->C en la segunda repetición del genotipo 3R que impide la unión de factores de trascripción USF-1 y USF-2. Los efectos de este polimorfismo sobre la eficacia o toxicidad de los fármacos que inhiben la actividad TS debe determinarse en estudios retrospectivos o prospectivos. Las toxicidades más frecuentes asociadas a la administración de capecitabina son el síndrome mano-pié, diarrea, náuseas, vómitos e irritación bucal. En general, la capecitabina es bien tolerada; los porcentajes de pacientes que tras el primer ciclo presentan toxicidades severas es de un 22% para el síndrome mano-pié, un 16% para diarrea y un 12% para irritación bucal. A los pacientes con toxicidad severa se les puede reducir la dosis en ciclos posteriores sin disminuir la eficacia. Lamentablemente, no hay forma de predecir qué pacientes van a experimentar una mayor toxicidad. Como los genotipos de TS son idénticos en los tumores primarios colorrectales y en los tejidos normales, se esperaría que los individuos con un genotipo 3R/3R tuvieran aumentada la expresión de la TS en tejidos normales, lo que protegería estos de la toxicidad de la capecitabina58. Un estudio pequeño y retrospectivo de pacientes con cáncer colorrectal tratados con distintos regímenes basados en el 5FU, mostró que la toxicidad grado 3 global eran un 36% inferior en individuos con polimorfismos 3R/3R en comparación con los pacientes 2R/2R59.Los efectos de los polimorfismos sobre el riesgo de toxicidad por el uso de la capecitabina en cáncer de mama en estadio temprano son desconocidos. Metiletentetrahidrofolato reductasa: la MTHFR también podría jugar un papel destacado con respecto a la toxicidad y eficacia de la capecitabina. Un polimorfismo de nucleótido único (SNP) en la posición 677 de la MTHFR, debido a una conversión de C en T, produce una enzima termolábil y de rápida degradación60. La frecuencia alélica de polimorfismos de la MTHFR varía con la localización geográfica y la raza. Se estima que la homocigosidad para TT está presente en aproximadamente el 10% de los norteamericanos61. Los homocigóticos para TT tienen niveles incrementados de metilentetrahidrofolato, un compuesto que estabiliza la unión del 5-FU a TS62, resultando en la formación del complejo ternario de 5-FU, TS y metilentetrahidrofolato. Nuestra hipótesis es que el aumento de la estabilización de este complejo ternario puede provocar un aumento tanto en la toxicidad como en la eficacia de la capecitabina. Un estudio retrospectivo realizado por Cohen et al63 apoya la hipótesis de que los polimorfismos en MTHFR pueden influir en la toxicidad y respuesta al tratamiento quimioterápico basado en fluoropirimidinas. En este estudio, 43 pacientes fueron tratados con uno de los siguientes

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 27 de 82
regímenes: i) capecitabina dos veces al día durante 2 semanas con una semana de descanso, ii) tegafur más uracilo tres veces al día durante 28 días con 7 días de descanso, iii) 5FU/LV administrados según el régimen de la clínica Mayo. Las tasas de respuesta y la frecuencia de las toxicidades se combinaron para los tres estudios: los homocigóticos de genotipo salvaje para MTHFR (C/C) tuvieron una tasa de respuesta del 47% comparada con una tasa de respuestas del 67% en los heterocigotos de MTHFR, y con un 100% de tasa de respuestas para los homocigotos T/T de MTHFR. Estas diferencias no fueron estadísticamente significativas debido al pequeño tamaño de la muestra. Cuando se analizaron los pacientes en función de la frecuencia de alelos mutantes, los pacientes con un alelo T tuvieron una tasa de respuesta del 77% comparada con un 55% de tasa de respuesta en pacientes con un alelo C presente. Esta comparación sí alcanzó significación estadística. MTHFR y la interacción con folato: los polimorfismos en MTHFR se han estudiado como posibles factores de riesgo en enfermedades tales como defectos del tubo neural en niños64, cáncer de colon65, y enfermedad cardiaca isquémica66. Los estudios epidemiológicos de MTHFR como factor de riesgo de enfermedades medían los niveles de folatos en los sujetos de la investigación para determinar si influían sobre el papel de los polimorfismos de MTHFR como factores de riesgo. El estudio de salud de los médicos65 mostró que el polimorfismo C677T de MTHFR protegía frente a la aparición de cáncer de colon solamente si los pacientes presentaban niveles adecuados de folatos. Christensen et al67 mostraron que las mujeres con genotipo TT del MTHFR presentaban un riesgo superior de tener un hijo con defecto del tubo neural, y que el riesgo aumentaba sustancialmente si la madre tenía deficiencia de folatos. En contraste, un estudio sobre los efectos del polimorfismo C677T en la toxicidad del metotrexato no mostró ninguna diferencia en pacientes suplementados o no con folatos. Este estudio no midió los niveles de folatos y usó los suplementos de folatos como un subrogado del estado de los niveles de folatos. Dihidropirimidina Deshidrogenasa: la deficiencia en dihidropirimidina deshidrogenasa (DPD) se ha identificado como la causa de reacciones amenazantes para la vida que aparecen raramente por la administración de fluoropirimidinas68,69. Los individuos con una deficiencia completa de la enzima tienen concentraciones significativamente elevadas de uracilo y timina en sangre y orina. Etienne et al70 se plantearon la posible utilidad del uso de la actividad de DPD como marcador de predicción del riesgo de toxicidad por fluoropirimidinas, mediante el estudio prospectivo de 185 pacientes tratadas con regímenes que contenían 5-FU. Encontraron en las pacientes una distribución normal de la actividad DPD, y ninguna correlación entre la misma y la toxicidad al 5-FU. Como conclusión, se parte de la hipótesis de que los polimorfismos en TS y MTHFR pueden predecir qué pacientes van a experimentar toxicidades y requerirán reducciones en la dosis de capecitabina. Así mismo los polimorfismos en TS y MTHFR podrían predecir qué pacientes no se beneficiarán de la dosis estándar de capecitabina. Especulamos que se encontrarán en el estudio los siguientes genotipos: i) genotipo 2R/2R de TS (toxicidad severa, beneficio del tratamiento adyuvante, necesidad de reducción de dosis); ii) genotipos 2R/3R de TS (toxicidad intermedia y beneficio intermedio del tratamiento adyuvante); y iii) genotipo 3R/3R de TS (poca o nula toxicidad y no beneficio del tratamiento adyuvante). 1.12 Cáncer de mama de tipo basal Los trabajos de diversos grupos de investigadores han consolidado la aparición de una clasificación tumoral del cáncer de mama basada en tres tipos de perfil genético: los tumores luminales (con receptores hormonales positivos), los tumores HER2 (con HER2neu positivo) y los tumores de tipo basal (con receptores hormonales y HER2neu negativos). Estos últimos,

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 28 de 82
también conocidos como los “triple negativo”, son el objeto del presente protocolo de investigación. Los tumores de tipo basal reciben este nombre por su perfil de expresión génica similar a la de un epitelio celular basal normal. Estas similitudes incluyen la ausencia de expresión del receptor de estrógeno y otros genes relacionados con el mismo y del receptor HER2. También comparten con el epitelio basal la sobre-expresión de citoqueratinas 5/6 y 17, de EGFR, así como de genes relacionados con la proliferación. Las mutaciones de p53 en tirosina son también características del tipo basal. Múltiples estudios independientes indican que los tumores de tipo basal presentan un pobre pronóstico. Sin embargo, su tratamiento es complicado, pues las pacientes con tumores triple negativo no son candidatas a tratamiento endocrino ni con trastuzumab. Sin embargo, este tipo de tumores es muy sensible a la quimioterapia. Investigadores del MD Anderson Cancer Center examinaron la quimiosensibilidad de este tipo de tumores71. El perfil genético se obtuvo mediante aspiración con aguja fina en 83 tumores de mama, previo al tratamiento neoadyuvante con paclitaxel y FAC. La respuesta patológica completa fue más frecuente entre los tumores de tipo basal, frente a los de tipo luminal. Este resultado se confirmó con un estudio de la Universidad de Carolina del Norte, en el que se clasificó el tipo de cáncer en 105 pacientes tratadas con AC y taxanos. El 27% de los tumores eran de tipo basal, , el 21% eran HER2 positivos, y el 52% eran de tipo luminal. Las mejores respuestas clínicas al tratamiento con quimioterapia se obtuvieron en las pacientes con tumores de tipo basal (86%, frente al 68% en las HER2 positivas y 60% en el tipo luminal)72. En paralelo, la tasa de respuesta patológica completa fue superior en las pacientes con tumores de tipo basal (30%, frente al 27% en las HER2 positivas y 13% en los tumores de tipo luminal). A pesar de presentar mayor sensibilidad a la quimioterapia, los tumores de tipo basal presentan un mal pronóstico. Con una mediana de seguimiento de 2,5 años en el estudio de la Universidad de Carolina del Norte, las pacientes con tumores de tipo basal presentaban una clara tendencia a recidivas tempranas. El motivo parece ser la falta de arsenal terapéutico para el tratamiento de pacientes con receptores hormonales negativos y HER2 negativo, por lo que, en caso de que quede enfermedad residual, el pronóstico es pobre. El Grupo cooperativo norteamericano B de Cáncer y Leucemia (CALGB), al hilo de este razonamiento, ha realizado una evaluación retrospectiva de sus estudios con quimioterapia, que revela que los beneficios de cada nueva generación de quimioterapia más agresiva, se centran sobre todo en las pacientes con receptores de estrógeno negativos. Esto sugeriría que las pacientes con tumores de tipo basal son las que mejor responderían a estrategias como aumentos de dosis o de intensidad de dosis de las antraciclinas, adición de taxanos a esquemas con antraciclinas, o aumentos en la densidad de dosis de los fármacos administrados. 1.13 Justificación para el estudio actual Los estudios adyuvantes representan el marco más óptimo para evaluar las nuevas propuestas de tratamiento de quimioterapia. La subjetividad de la evaluación de la tasa de respuesta observada en los ensayos metastásicos se ve reemplazada por la objetividad de las variables empleadas en los mismos (supervivencia libre de enfermedad y, finalmente, supervivencia global). Como se ha comentado, la mayoría de los estudios relevantes que se llevan a cabo en estos momentos en adyuvancia, están utilizando protocolos secuenciales, con la idea de poder administrar la mayor dosis posible de cada fármaco disminuyendo la adición de toxicidad que se puede producir al dar diversos quimioterápicos simultáneamente. En esta línea, el diseño del estudio CIBOMA/2004-01 destaca por los siguientes motivos:
- Va dirigido hacia un tipo de tumor de pobre pronóstico, el tipo basal o triple negativo. Se postula que este tipo de tumor es el que más puede beneficiarse de los tratamientos

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 29 de 82
con quimioterapia. Además, estas pacientes no disponen de la posibilidad de recibir tratamiento complementario con hormonoterapia o trastuzumab.
- Se estudia la hipótesis del posible beneficio de una tratamiento de quimioterapia más prolongado (6 ciclos de quimioterapia adyuvante estándar? 8 ciclos de capecitabina).
- Se usa como fármaco experimental la capecitabina, que ha demostrado ser uno de los más eficaces y seguros en el tratamiento del cáncer de mama metastásico.
- La capecitabina es un fármaco que se administra por vía oral, lo que permitirá prolongar el tratamiento adyuvante con quimioterapia adyuvante hasta 12 meses, sin que afecte a la calidad de vida de las pacientes tratadas.
2 OBJETIVOS Y PROPÓSITO DEL ESTUDIO 2.1 Objetivo Primario: Comparar la supervivencia libre de enfermedad a 5 años después de terapia de mantenimiento con 8 ciclos de capecitabina (X) frente a observación, en pacientes con cáncer de mama operable, receptores hormonales y HER2 negativos, que han recibido un tratamiento quimioterápico adyuvante estándar. 2.2 Objetivos secundarios:
?? Comparar la Supervivencia Global (SG) entre los dos grupos mencionados anteriormente.
?? Comparar la toxicidad entre los 2 grupos mencionados anteriormente.
2.3 Objetivos terciarios: ?? Determinar el efecto del tratamiento con capecitabina sobre la aparición de amenorrea
en mujeres premenopáusicas y duración de la misma. ?? (En centros seleccionados): Determinar la existencia de polimorfismos de la enzima
timidilato sintasa (TS) y en la enzima metilentetrahidrofolato reductasa (MTHFR), y comprobar su asociación con la toxicidad y eficacia del tratamiento con capecitabina.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 30 de 82
3. SELECCIÓN DE LOS PACIENTES 3.1 Criterios de inclusión
1. Se debe obtener y documentar el consentimiento informado por escrito antes de iniciar los procedimientos específicos del protocolo, incluida la cooperación esperada de las pacientes en el tratamiento y el seguimiento, de acuerdo con las normas ICH de Buena Práctica Clínica. La paciente deberá firmar un consentimiento informado para envío de su muestra tumoral a laboratorio central. Posteriormente, si la paciente es elegible, deberá firmar un consentimiento para su participación en el ensayo clínico.
2. Cáncer de mama histológicamente demostrado (examen histológico del tumor: adenocarcinoma invasivo).
3. Pacientes con afectación axilar ganglionar ipsilateral. En caso de utilizarse la técnica del ganglio centinela, el mismo contará como ganglio extirpado y afectado. Se admitirán las pacientes que se puedan clasificar en los siguientes grupos (AJCC, 2002):
- pN1a: Metástasis en 1 a 3 ganglios axilares. - pN2a: Metástasis en 4 a 9 ganglios axilares (al menos un depósito tumoral > 0,2
cm). - pN3a: Metástasis en 10 ó más ganglios axilares (al menos un depósito tumoral
> 0,2 cm). No serían elegibles las pacientes con diagnóstico pN3a con metástasis en ganglios infraclaviculares.
Nota: Pacientes sin afectación ganglionar axilar (N0) son también elegibles, siempre que el tumor primario mida más de 2 cm de diámetro. En caso de utilizarse la técnica del ganglio centinela, no será necesario realizar linfadenectomía.
4. Mujeres en régimen ambulatorio de edad? 18 años. 5. Índice del estado funcional de Karnofsky ? 80 (ECOG 0,1).
3.2 Criterios de exclusión
1. La paciente no ha sido sometida a tratamiento quirúrgico definitivo para el cáncer de mama operable (T1-3, M0) bien mastectomía, o cirugía conservadora de la mama; o los márgenes de la muestra extraída de la cirugía definitiva son histológicamente positivos para adenocarcinoma invasivo o carcinoma ductal in situ (DCIS). El carcinoma lobular in situ no se considera un margen positivo.
2. La paciente no ha recibido al menos 6 ciclos de quimioterapia adyuvante previa con esquemas que contengan antraciclinas y/o taxanos (docetaxel o paclitaxel). Los tratamientos permitidos se encuentran disponibles en el apéndice 1. Cualquier otro esquema de tratamiento deberá ser aprobado por los coordinadores médicos del estudio.
3. En la linfadenectomía, extirpación de menos de 6 ganglios (para las pacientes con biopsia del ganglio centinela, si este es positivo, cuenta como extirpado).
4. Terapia anterior con antraciclinas o taxanos (paclitaxel, docetaxel) para cualquier neoplasia diferente del cáncer de mama que está siendo tratado.
5. Para pacientes que no reciben radioterapia, el intervalo entre el día 1 del último ciclo de quimioterapia adyuvante y la entrada en el ensayo clínico es superior a 8 semanas (2 meses). Para las pacientes en las que está indicada la administración de radioterapia adyuvante, el intervalo entre la última sesión y la entrada en el estudio es superior a 4 semanas (1 mes).
6. Pacientes con tumores con receptores de estrógeno, progesterona y HER2 desconocidos, o positivos.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 31 de 82
7. Pacientes embarazadas o en período de lactancia. Pacientes con potencial de fertilidad y un resultado desconocido o positivo en la prueba de embarazo en orina o suero, realizada durante los 14 días anteriores a la asignación de tratamiento.
8. Mujeres en edad fértil que no desean usar un método anticonceptivo fiable y apropiado. Las mujeres postmenopáusicas deben haber permanecido amenorreicas durante al menos 12 meses para ser consideradas como no fértiles.
9. Diagnóstico de cáncer de mama bilateral invasivo. 10. Enfermedad cardiaca clínicamente relevante, tal como insuficiencia cardiaca
congestiva o enfermedad arterial coronaria sintomática, arritmias cardiacas no controladas con tratamiento o historia anterior de infarto de miocardio durante los 12 meses anteriores a la inclusión en el estudio, o hipertensión no controlada.
11. Historia de trastornos neurológicos o psiquiátricos significativos, incluidos trastornos sicóticos, demencia o ataques que impedirían a la paciente entender y otorgar el consentimiento informado, interferirían en el cumplimiento de la pauta del fármaco en estudio.
12. Infección activa no controlada, u otras enfermedades o patologías médicas graves, tales como ulcera péptica activa, diabetes mellitus inestable.
13. Presencia de anomalías en cualquiera de los parámetros de laboratorio, tal y como se definen a continuación: (en los 14 días anteriores a la asignación de tratamiento):
?? Hemoglobina < 10 mg/dl; recuento absoluto de neutrófilos < 1,5 x 109/l; recuento de
plaquetas < 100x 109/l. ?? ASAT (SGOT) y ALAT (SGPT) > 2,5 LSN; Fosfatasa alcalina > 5 LNS; Bilirrubina
total > 2.0 LSN. Las pacientes con valores de ASAT y/o ALAT > 1,5 x LNS asociados con un nivel de fosfatasa alcalina > 2,5 x LNS no se seleccionarán para el estudio.
?? Insuficiencia renal severa, definida como aclaramiento de creatinina calculado inferior a 30 ml/min, según la fórmula de Cockroft y Gault (véase apéndice 8) o creatinina sérica > 1,5 LSN.
14. Historia anterior o actual de neoplasias distintas al cáncer de mama, a excepción de: ?? Cáncer de piel no melanoma, carcinoma de cérvix uterino in situ u otro tipo de
tumor tratados de forma curativa y sin indicios de enfermedad durante, al menos, diez años.
?? Carcinoma ductal in situ (DCIS) ipsilateral de mama. ?? Carcinoma lobular in situ (LCIS) de mama.
15.Hipersensibilidad conocida a capecitabina, fluorouracilo o a cualquiera de los excipientes. 16. Pacientes con falta de integridad física del tracto gastrointestinal superior o que
manifiesten síndrome de mala absorción, o imposibilidad de tomar medicación por vía oral.
17. Tratamiento previo con capecitabina o con infusión continua (> 24 h) con 5-FU, o con otras fluoropirimidinas orales, tales como eniluracil/5-FU, uracilo/tegafur, S1 o emitefur.
18. Transfusiones de sangre o administración de factores de crecimiento para ayudar a la recuperación hematológica en las 2 semanas previas al comienzo del tratamiento del estudio.
19. Tratamiento hormonal en los 10 días previos al comienzo del tratamiento del estudio. 20. Posibilidad de reacción severa no prevista a terapia con fluoropirimidina, con o sin
probada deficiencia de dihidropirimidina dehidrogenasa (DPD). 21. Transplantes alográficos de órganos que requieran terapia inmunosupresora. 22. Pacientes con tratamiento anticoagulante con derivados de cumarina. 23. Pacientes en tratamiento con sorivurina o sus análogos químicamente relacionados,
como la brivudina.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 32 de 82
24. Tratamiento concomitante con otros fármacos en investigación. Participación en otro ensayo clínico con cualquier fármaco en investigación no comercializado durante los 30 días previos a la inclusión en el estudio.
25. Tratamiento concomitante con cualquier otra terapia anticancerosa. 26. Pacientes no accesibles para el tratamiento y el seguimiento. Las pacientes registradas
en este ensayo deberán ser tratadas y seguidas en el centro participante, que podría ser el centro del investigador principal o co-investigador.
27. Pacientes que no puedan o quieran cumplir con el protocolo, considerando la duración completa del estudio.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 33 de 82
4. ESTRATIFICACIÓN Y ASIGNACIÓN ALEATORIA DE BRAZO DE TRATAMIENTO Los investigadores participantes serán responsables de comprobar que cada nueva paciente cumple con los criterios de inclusión del estudio, es informada apropiadamente acerca del mismo, lee y comprende las Hojas de Información al paciente, y firma y fecha de su puño y letra el consentimiento informado para participar en el estudio. 4.1 Calendario de registro y asignación aleatoria en el estudio Las pacientes potencialmente elegibles, deberán firmar el consentimiento informado para envío de la muestra al laboratorio central al comienzo del tratamiento con quimioterapia adyuvante estándar. La recepción de la muestra coincide con el registro de la paciente en el estudio. Los exámenes a realizar en la muestra son : estado de receptores de estrógeno y progesterona y de HER2. El investigador recibirá la respuesta desde el laboratorio central, acerca de si la paciente puede participar en el estudio, antes del final del tratamiento con quimioterapia estándar. Si la paciente es elegible, deberá firmar un nuevo consentimiento informado para aceptación de su participación en el estudio. Para las pacientes que no reciban radioterapia adyuvante, el intervalo entre el día 1 del último ciclo de quimioterapia adyuvante y la asignación aleatoria en el estudio debe ser como máximo 8 semanas (2 meses). Para las pacientes en las que está indicada la administración de radioterapia adyuvante, el intervalo entre la última sesión y la asignación aleatoria al brazo de tratamiento del estudio debe ser como máximo 4 semanas (1 mes). El coordinador administrativo del estudio deberá registrar a todas las pacientes seleccionadas, antes del inicio del tratamiento del estudio. Las pacientes que no se hayan registrado antes de la administración del primer tratamiento no serán aceptadas posteriormente para participar en el estudio. Para las pacientes asignadas al brazo de capecitabina, no deberán transcurrir más de 8 días desde la fecha de asignación aleatoria de tratamiento hasta la fecha de inicio del primer ciclo de mantenimiento con capecitabina de mantenimiento. El monitor del estudio notificará al investigador por fax y/o correo electrónico, en el plazo de un día laborable, el número de paciente del estudio y el brazo de tratamiento que se le ha asignado aleatoriamente. 4.2 Procedimiento de asignación aleatoria del tratamiento Las hojas de registro se deberán enviar por fax o correo electrónico al coordinador administrativo del estudio (Departamento Central de Operaciones de CIBOMA):
CIBOMA Teléfono: + 34 916592870 Fax: +34 916510406 [email protected]
La asignación aleatoria de tratamiento se realizará una vez comprobado que la paciente estaba previamente registrada, y es elegible. 4.3 Grupos de tratamiento Mediante el proceso de distribución aleatoria, se asigna a las pacientes a uno de los dos grupos siguientes:
?? Brazo A: tratamiento de mantenimiento con capecitabina. ?? Brazo B: observación

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 34 de 82
4.4 Estratificación Cada paciente seleccionada se asignará de forma aleatoria a un brazo de tratamiento según un bloque específico (centro, tratamiento adyuvante previo con antraciclinas vs. antraciclinas y taxanos, número de ganglios afectados (0 vs.1-3 vs. ?4), para que reciba Xx8 u observación.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 35 de 82
5. TRATAMIENTO DE LOS PACIENTES Brazo experimental: capecitabina 1.000 mg/ m2 dos veces al día durante 14 días, seguidos de un periodo de descanso de 7 días, durante 8 ciclos. El tratamiento en estudio es capecitabina (Xeloda®). Para el propósito del estudio, la medicación se definirá como la administración de capecitabina en el brazo de tratamiento durante todo el tratamiento activo. Xeloda® (capecitabina) es un carbamato de fluoropirimidina no citotóxica que, administrado por vía oral, actúa como un precursor del citotóxico 5-fluorouracilo (5-FU). La capecitabina se activa a través de varios pasos enzimáticos. La enzima responsable de la conversión final a 5-FU, la timidina fosforilasa, se encuentra en tejidos tumorales así como en tejidos normales aunque con niveles generalmente más bajos.
Brazo Control: observación. 5.1 Aportación de los fármacos La compañía farmacéutica Roche aportará el fármaco Xeloda? como medicación comercial, en unidades suficientes para el tratamiento de las pacientes del estudio de forma gratuita. La medicación del estudio será entregada por el Servicio de Farmacia a los investigadores del estudio, quienes deberán dispensarla a las pacientes y asegurar el cumplimiento. Xeloda? se presenta como cajas de comprimidos con cubierta pelicular: ?? Xeloda? 500 mg: 120 comprimidos con cubierta pelicular (12 blísters de 10 comprimidos).
Los comprimidos no están hendidos, y no debieran partirse. Xeloda? no se debe almacenar a una temperatura superior a los 30ºC. Se proporcionará a los investigadores principales en cada centro una tabla de registro de temperaturas, que deberá completarse en el lugar de almacenamiento del fármaco en estudio al menos una vez por semana. Se informará a las pacientes adecuadamente acerca de las condiciones de almacenamiento de Xeloda? . 5.2 Administración de capecitabina Las dosis de quimioterapia con Xeloda? se administrará en función del área corporal, y ajustando la dosis al alza o a la baja, hasta la aproximación más cercana a la suma total de 500. Se ruega remitirse a la tabla del apéndice 9, para determinar el redondeo apropiado de dosis en función de la superficie corporal. Si se producen variaciones del área corporal durante el estudio, se asumirá que el área corporal permanece aproximadamente constante, es decir, no se realizarán ajustes por cambios en el peso corporal a lo largo del estudio. Capecitabina Se administra a una dosis de 1000 mg/m2, dos veces al día (mañana y noche); equivalente a una dosis diaria total de 2.000 mg/m2. Un ciclo consiste en la administración durante 14 días, seguido de 7 días de descanso. A discreción de investigador y paciente, los ciclos se pueden calcular de dos formas: la primera pauta, permite que la primera dosis de cada ciclo se administre en la mañana del día 1, y la última dosis, en la tarde del día 14, seguido de un periodo de 7 días de descanso. De esta forma, el ciclo consiste en la administración de 28 dosis en un periodo de 14 días de calendario. La segunda pauta permite que la primera dosis de cada ciclo se administre en la tarde del día 1, y la última dosis en la mañana del día 15, seguido de un periodo de descanso de 6,5 días. Esto proporciona un total de 28 dosis por ciclo, en 15 días de calendario. Se puede usar cualquiera de estas pautas, siempre y cuando la paciente reciba 28 dosis administradas en días consecutivos. La paciente no precisa seguir la misma pauta durante todos los ciclos, y puede escoger cambiar de pauta a partir de un determinado ciclo. Por

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 36 de 82
ejemplo, en el ciclo 1, la paciente acude a consulta al hospital por la mañana, y comienza el ciclo por la tarde. Toma la última dosis en la mañana del día 15. En el segundo ciclo, la paciente toma la primera dosis en la mañana del día 1, y la última en la tarde del día 14. Se deberá registrar en el CRF la pauta seguida en cada ciclo. Las dosis de mañana y tarde deben distanciarse, al menos, en 12 horas una de otra. Se deberá ingerir la dosis con 200 ml de agua dentro de los 30 minutos posteriores a una comida. Por ejemplo, una paciente con superficie corporal de 1,6 m2, debería recibir 1.600 mg. Se redondea al alza para administrar 4 comprimidos de 500 mg. Se proporcionará a la paciente medicación suficiente para completar las 28 dosis en cada ciclo de 21 días. Se administrarán 8 cic los de tratamiento. Dosis de inicio en pacientes con insuficiencia renal moderada Las pacientes con insuficiencia renal moderada en el momento basal, definida como aclaramiento de creatinina entre 30-50 ml/min (calculado según la fórmula de Cockroft y Gault, apéndice 8), comenzarán con una reducción en la dosis del 75%, es decir, en lugar de 100 mg/m2 dos veces al día, recibirán 750 mg/m2 dos veces al día. 5.3 Medicación concomitante (sólo aplicable a pacientes del brazo de tratamiento con capecitabina) Fenitoína Se ha informado de un aumento en las concentraciones plasmáticas de fenitoína, durante la administración concomitante con capecitabina. No se han realizado estudios formales de interacción entre ambos fármacos. Las pacientes que reciban fenitoína de forma concomitante con capecitabina, deberán ser monitorizadas regularmente para controlar sus niveles plasmáticos de fenitoína, y los síntomas asociados. Halopurinol Se han observado interacciones entre halopurinol y 5-FU, con una posible disminución de la eficacia del 5-FU. Se deberá evitar la administración conjunta de halopurinol y capecitabina. Metronidazol No se permite su administración concomitante con capecitabina en el presente estudio. Cimetidina Se ha informado que el tratamiento previo durante 4 semanas con cimetidina, aumenta las concentraciones plasmáticas de fluorouracilo, tras administración intravenosa y oral a 6 pacientes. El efecto se debió, probablemente a una combinación de la inhibición de enzimas hepáticas y reducción del flujo sanguíneo en hígado. No se ha observado este efecto tras la administración de dosis únicas de cimetidina a 5 pacientes, o en el pretratamiento durante 1 semana en 6. Se aconseja precaución si se debe administrar de forma concomitante cimetidina y capecitabina a una paciente del estudio. Factores de crecimiento hematopoyético Se pueden administrar de acuerdo a las pautas habituales en cada centro, para el tratamiento de la neutropenia febril. No se puede administrar como tratamiento profiláctico primario ni secundario. Se anotará la administración de los factores de crecimiento hematopoyético en la historia clínica y en el CRD. Se deberá interrumpir la administración de factores de crecimiento al menos 48 horas antes del inicio del siguiente ciclo de capecitabina. Laxantes Se debe evitar la administración de fármacos con propiedades laxantes, por el riesgo de exacerbación de la diarrea.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 37 de 82
5.4 Ajustes de dosis permitidos Las toxicidades se clasificarán utilizando los criterios comunes de toxicidad del NCI, versión 3.0 (http://ctep.cancer.gov/reporting/ctc.html). Están previstas dos reducciones de dosis posibles en caso de una toxicidad hematológica y/o no hematológica severa de la forma que se indica a continuación: Fármaco De (dosis inicial) A (dosis reducida-75%) A (dosis reducida-50%) Capecitabina 2000 mg/m2 (1000
mg m2 2 veces al día) 1500 mg/m2 (750 mg m2
2 veces al día) 1000 mg/m2 (500 mg m2 2
veces al día) Una vez que se reduzca la dosis, no deberá incrementarse en ningún momento posterior. Las interrupciones en el tratamiento con capecitabina se considerarán como días de tratamiento perdidos, y se mantendrá el plan de tratamiento inicial. Las dosis perdidas debidas a interrupciones en el tratamiento por toxicidad, no serán reemplazadas (véase apéndice 9). Si la paciente experimenta varias toxicidades de diferente graduación o intensidad al mismo tiempo, se aplicará la reducción de dosis mayor aplicable. Si el valor de aclaramiento de la creatinina disminuye durante el estudio a un valor < 50 ml/min, debido a un aumento en la creatinina sérica o a una disminución en el peso corporal, este cambio no es motivo, en sí mismo, para una reducción de dosis. Las reducciones de dosis durante el tratamiento deben basarse en acontecimientos adversos. Con respecto a los acontecimientos adversos que son aparentes en el momento basal, las modificaciones de dosis se aplicarán de acuerdo al correspondiente cambio en la gradación de la toxicidad, si el investigador lo considera apropiado. Por ejemplo, si una paciente presenta astenia grado 1 en el momento basal, que aumenta a grado 2 durante el tratamiento, la diferencia a considerar es el aumento de 1 grado en la toxicidad, a la hora de realizar modificaciones de dosis. En el caso de toxicidades que el investigador considere improbable que se conviertan en graves o amenazantes para la vida (por ejemplo, alopecia, alteraciones del gusto), no se deberá interrumpir ni reducir la dosis del fármaco. Tampoco se requieren reducciones o interrupciones de dosis en caso de anemia (no hemolítica), si he trata satisfactoriamente mediante trasfusiones. En caso de toxicidad grado 1, o de alopecia, el tratamiento continuará a la dosis inicial sin interrupciones. Se aplicarán los criterios siguientes con respecto a la reducción de dosis o interrupción del tratamiento con capecitabina por toxicidad:

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 38 de 82
Pauta de reducción de dosis para la monoterapia con capecitabina- Toxicidad no hematológica Grados 2 y 3 Grado 4 -1ª aparición Interrumpir hasta que remita a grado 0-1;
continuar la administración de capecitabina al 75% de la dosis inicial, añadiendo profilaxis si es posible.
Interrumpir permanentemente el tratamiento, a menos que el investigador considere que
continuarlo es la mejor opción terapéutica para la paciente, en cuyo caso se retomará al 50% de la dosis
original. -2ª aparición Interrumpir hasta que remita a grado 0-1;
continuar la administración de capecitabina al 50% de la dosis inicial.
-3ª aparición Interrumpir permanentemente el tratamiento, a menos que el investigador considere que continuarlo es la mejor opción terapéutica para la paciente.
La capecitabina puede inducir diarrea, en ocasiones severa. En pacientes que reciben capecitabina en monoterapia, la mediana de tiempo hasta aparición de diarrea grado 2-4 es de 31 días, y la duración mediana de las diarreas grado 3 o 4 fue de 4,5 días. Las pacientes con diarrea severa deberán ser monitorizadas atentamente, y en caso de deshidratación, se garantizará el aporte de fluidos y electrolitos. En caso de diarrea grados 2, 3 ó 4, de debe interrumpir de inmediato la administración de la capecitabina, hasta su resolución o disminución a grado 1. Los siguientes episodios de diarrea grado 2 o superior, requieren una reducción de dosis de capecitabina, como se establece en la tabla anterior. Se deberá comenzar el tratamiento con fármacos antidiarreicos estándar, como loperamida, tan pronto como sea posible. No se deberá reiniciar al tratamiento con capecitabina hasta que se haya resuelto la diarrea a grado 0-1, habiéndose administrado la última dosis de loperamida al menos 24 horas antes. Náuseas /Vómitos grado ? 2 Se debe proporcionar a las pacientes medicación antiemética para auto administración en caso de náuseas y vómitos, cuando estos ocurren en casa. Se recomienda la metoclopramida +/- antagonistas 5-HT3 para las náuseas inducidas por capecitabina. Se iniciará la terapia profiláctica y secundaria apropiadas cuando se hayan producido náuseas y vómitos. Si recurren las náuseas o vómitos, a pesar de la profilaxis, se realizará una modificación de dosis de acuerdo a lo establecido en la tabla correspondiente. Toxicidad cutánea: síndrome mano-pié grado 2/3 El síndrome mano-pié (eritrodisestesia palmo plantar o eritema inducido por quimioterapia) se considera toxicidad cutánea. Si se produce un síndrome mano-pié de grado 2 ó3, se debe interrumpir la administración de capecitabina hasta la resolución o disminución a grado? 1. Las disminuciones posteriores de dosis se resumen en la tabla de este mismo apartado. El síndrome mano-pié debe ser tratado sintomáticamente (por ejemplo, se recomienda el uso de emolientes). Se ha reportado un posible beneficio del uso de la vitamina B6 p piridoxina73,74, y se permite como tratamiento sintomático o profilaxis secundaria del síndrome mano-pié. 5.4.1 Advertencias y precauciones La capecitabina se administra de forma ambulatoria, y en ciertas circunstancias, efectos adversos como la diarrea pueden rápidamente convertirse en eventos graves. En el caso en que una paciente experimente cualquier toxicidad entre dos visitas programadas, se debe instruir a la

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 39 de 82
paciente para que avise a su médico tan pronto como pueda, para que éste la instruya acerca de posibles tratamientos. Es importante instruir a las pacientes para que interrumpan el tratamiento con capecitabina tan pronto se produzcan toxicidades de grado 2 de los eventos más frecuentes, como diarrea, síndrome mano-pié y estomatitis. La capecitabina está contraindicada en pacientes con insuficiencia renal grave (aclaramiento de creatinina por debajo de 30 ml/min (apéndice 8). Las pacientes con insuficiencia renal grave no son elegibles para este estudio. Si la misma se produjera durante el tratamiento del estudio, se interrumpirá de inmediato. Se ha informado que en pacientes tratados con capecitabina, y que presentaban insuficiencia renal moderada (aclaramiento de creatinina 30-50 ml/min), la incidencia de efectos adversos de grado 3 ó 4 aumenta. Si se incluye en el estudio una paciente con insuficiencia renal moderada, se comenzará el tratamiento con la dosis correspondiente al 75%. En pacientes con insuficiencia renal leve (aclaramiento de la creatinina de 51-80 ml/min), no se realizará ningún ajuste de la dosis basal, pero se vigilará la aparición de toxicidad grado 2, 3 ó 4, para realizar los ajustes de dosis establecidos por protocolo. Se han reportado eventos de cardiotoxicidad asociados a tratamiento con pirimidinas fluorinadas (incluyendo la capecitabina), que incluyen el infarto de miocardio, angina, disrritmias, shock cardiogénico, muerte súbita y cambios electrocardiográficos. Estos efectos adversos pueden ser más comunes en pacientes con antecedentes de enfermedad coronaria arterial. En pacientes que experimenten elevaciones de bilirrubina de > 3 LSN, o elevación de las enzimas hepáticas transaminasas de > 2,5 LSN, relacionadas con el tratamiento, se interrumpirá la administración de capecitabina. Se reanudará la misma, cuando se alcancen niveles de bilirrubina<3.0 LSN, o de enzimas hepáticas transaminasas de < 2,5 LSN. Los pacientes mayores de 80 años corren más riesgo de experimentar toxicidad grado 3 ó 4, en particular la diarrea, náusea, síndrome mano-pié y vómitos. 5.5 Contabilidad de la medicación La persona responsable de dispensar la medicación deberá mantener un registro adecuado del fármaco suministrado durante el estudio. Dicho registro incluye la fecha en la que se ha recibido la medicación del estudio, así como las fechas en que se ha dispensado a la paciente, el número de comprimidos administrados en cada visita, y el número de comprimidos devueltos en la siguiente visita de cada paciente. 5.6 Quimioterapia adyuvante estándar La quimioterapia adyuvante estándar previa a la randomización consistirá en un mínimo de 6 ciclos de combinaciones de antraciclinas con otros antineoplásicos, o antraciclinas y taxanos. En el apéndice 1 se encuentra la tabla de regímenes de quimioterapia adyuvante previa permitidos en este estudio, y que las pacientes deben haber completado para ser elegibles para el estudio CIBOMA/2004-01.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 40 de 82
6. EVALUACIÓN DE LA EFICACIA 6.1 Variable principal: evaluación de la eficacia del tratamiento. Todas las pacientes randomizadas se incluirán en un análisis de intención de tratar. La evaluación primaria de la eficacia consistirá en la comparación de la supervivencia libre de enfermedad (SLE) entre los dos grupos de tratamiento a los 5 años. La SLE se define como el intervalo desde la fecha de randomización hasta la fecha de la recurrencia local, regional o metastásica o la fecha del segundo cáncer primario (exceptuando el carcinoma de células escamosas o células basales de piel, carcinoma in situ de cérvix uterino o carcinoma lobular in situ de mama) o la muerte por cualquier causa, según la circunstancia que se manifieste en primer lugar. Si se suspende el tratamiento del estudio (cualquiera que sea el motivo), se realizarán los análisis de supervivencia libre de enfermedad y supervivencia de la paciente, según el análisis de intención de tratar. Las variables que se utilizarán para medir la eficacia del tratamiento son: 6.1.1 Recurrencia objetiva: Cualquier indicio clínico o radiológico de recurrencia tumoral, incluido el sistema nervioso central. Se debe obtener la prueba histológica o citológica del fracaso, si es posible. La aparición de cualquier indicio de enfermedad maligna se deberá detallar en los diagramas. Se realizará el seguimiento para la supervivencia.
6.1.2 Recurrencia local: Se define como reaparición del tumor en la cicatriz quirúrgica de la mama, mama ipsilateral (en el caso de cirugía conservadora) o indicios de tumor en la pared torácica anterior ipsilateral (mastectomía) o en la piel o tejidos blandos dentro del área local. Preferiblemente realizar prueba histológica o citológica confirmatoria. 6.1.3 Recurrencia regional: Se define como reaparición del tumor en la cicatriz axilar, áreas ganglionares ipsilaterales (axilares, supraclaviculares, mamarias internas e infraclaviculares), así como en la piel o tejidos blandos dentro del área regional. Preferiblemente realizar prueba histológica o citológica confirmatoria. 6.1.4 Recurrencia a distancia: Se define como los indicios del tumor más allá del nivel locorregional, según se ha definido anteriormente. Incluye las siguientes áreas: 1) ganglios linfáticos no incluidos en las áreas mencionadas anteriormente (es decir, axila contralateral, paratraqueal, etc) 2) piel no incluida en las áreas definidas anteriormente 3) hígado 4) pulmón 5) huesos 6) sistema nervioso central 7) mama contralateral 8) otras localizaciones no definidas anteriormente Preferiblemente realizar prueba histológica o citológica, especialmente en las lesiones solitarias. Las gammagrafías óseas positivas se deben correlacionar con la radiografía ósea.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 41 de 82
Los nódulos pulmonares múltiples en la radiografía de tórax, los nódulos hepáticos múltiples en la ecografía hepática o TAC, las lesiones óseas líticas o blásticas múltiples o focos de hipercaptación múltiples en la gammagrafía ósea serán aceptables sin correlación patológica. 6.1.5 Segundo tumor primario: Se define como cualquier otro tumor demostrado histopatológicamente, incluido segundo cáncer de mama primario invasivo en la mama ipsilateral o contralateral. Se excluyen el cáncer de piel de tipo no melanoma, el carcinoma de cérvix uterino in situ y el carcinoma de mama in situ (LCIS/DCIS). 6.2 Variable secundaria: Supervivencia Global a los 5 años La supervivencia se determinará desde la fecha de la randomización hasta la fecha en que se produzca el fallecimiento por cualquier motivo. 6.3 Variable secundaria: evaluación de la seguridad del tratamiento. 6.3.1 Seguridad clínica Las siguientes pruebas se realizarán antes de y/o en días específicos durante la terapia: ?? Historia completa de enfermedades malignas y no malignas, incluidos los antecedentes
cardiacos. ?? Examen clínico completo, signos vitales (presión arterial, pulso, temperatura), talla, peso,
evaluación de cualquier toxicidad residual debida a la terapia anterior, evaluación del estado funcional según el índice de Karnofsky.
?? Radiografía de tórax. ?? Acontecimientos adversos: se realizará regularmente una evaluación de cada paciente para
determinar los posibles acontecimientos adversos según la escala del NCI CTC (v.3.0). ?? Determinaciones de laboratorio: las siguientes pruebas se realizarán antes de y en días
específicos durante la terapia: ?? Hematología: Recuento leucocitario, neutrófilos y plaquetas, hemoglobina ?? Bioquímica: Parámetros habituales de funciones hepática y renal (creatinina o aclaramiento). ?? En caso de dolor óseo, elevación de fosfatasa alcalina o elevación de marcadores tumorales, debe realizarse una gammagrafía ósea. En cualquier caso, se recomienda la realización de una gammagrafía ósea que sirva de imagen basal para posibles controles ulteriores. Las toxicidades que no se puedan clasificar por medio de los criterios de toxicidad común del NCI, se definirán de la siguiente manera:
a. leve (asintomática) b. moderada (sintomática, pero sin interferir significativamente en la función) c. grave (interfiere significativamente en la función) d. amenazante para la vida

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 42 de 82
6.4 Variable terciaria (en poblaciones específicas de pacientes): Para comprobar prospectivamente el efecto de los polimorfismos de TS y MTHFR asociados a la eficacia y toxicidad de la administración de capecitabina, se obtendrá una muestra de sangre de cada paciente antes del inicio del tratamiento. La muestra será procesada para obtención de células de la serie blanca y plasma. Una vez en el laboratorio, se realizará la extracción de los glóbulos blancos a partir de la muestra y se aislará el ADN de las células mediante el mini-kit QIAamp para purificación de ADN de Qiagen. La región del promotor de TS se amplificará mediante PCR usando los cebadores previamente descritos en la literatura. Los productos de la reacción se resolveran mediante gel de agarosa al 1%. Se secuenciarán los productos del PCR de diferentes longitudes, y se identificarán los fragmentos representativos de 2R (220 pb) y 3R (248 pb). Estos productos se usarán como marcadores de tamaño y se incluirán como controles de experimentos posteriores. Se secuencierán los productos 3R para polimorfismos descritos en la literatura. Las muestras identificadas como alelos 3R serán clonadas usando un vector T de clonación (Invitrogen) y el inserto se secuenciará con cebadores del vector usando kits de secuenciación y un analizador genético de electroforesis capilar de Applied Biosystems. El programa GeneTools de Biotool se usará para analizar los datos de la secuenciación. Se hallará el genotipo de los pacientes en la región de 6 pb en el extremo 3´UTR para polimorfismos, siguiendo el método modificado de Ulrich. Se usarán los siguientes cebadores: 5´CAAATCTGAGGAGCTGAGT3´y 5´CAGATAAGTGGCAGTACAGA3´. Las condiciones del PCR serán las siguientes: 1 ciclo a 94ºC durante 5 minutos; 30 ciclos a 94ºC durante 30 segundos; 57ºC durante 50 segundos; 74ºC durante 45 segundos, y 1 ciclo de 5 minutos a 72ºC. Los fragmentos se digerirán con DraI y se separarán mediante gel de agarosa al 3%. La delección de 6 pb produce un fragmento de 152 pb y los tipos salvajes producen fragmentos de 70 y 88 pb. Para determinar los genotipos de MTHFR, se analizarán los polimorfismos de nucleótido único en las posiciones de nucleótido 677 T/C y 1298 A/C con las bases de datos de identificación de polimorfismos de nucleótido único rs1801133 y rs1801131 (Genbank ref. mRNA NM 005957), respectivamente, usando el pirosecuenciador PSQ HSA96. Se usarán los siguientes cebadores:
- Cebador directo de C677T: 5´CTAGGAAGGTGCAAGATCAGAG; - Cebador inverso de C677T: 5´Biotina-GTGTCAGCCTCAAAGAAAAG - Cebador de secuenciación de C677T: GGTGTCTGCGGGA. - Cebador directo de A1298C: 5´AACTCACTTTGTGACCATTCC - Cebador inverso de A1298C: 5´Biotina-GGGGAGCTGAAGGACTACTA - Cebador de secuenciación de A1298C: 5´AAGACTTCAAAGACACT
La cadena simple biotinilada de ADN de los polimorfismos de MTHFR y sus regiones cercanas se generarán usando técnicas estándar de PCR y secuenciando los genotipos con el pirosecuenciador según instrucciones del fabricante. Los cebadores del PCR se diseñarán usando el programa Primer3 (Whitehead) y los cebadores de secuenciación se diseñarán mediante el programa Pyrosequencing Inc. Todos los cebadores se sintetizarán por encargo a Qiagen Genomics. Para la determinación de deficiencias en la DPD, se evaluarán los niveles en sangre de timina y uracilo. Se eliminarán las proteínas del plasma mediante ácido tricloroacético, y se neutralizará con tri-n-octilamina en 1,1,2-triclorofluoroetano. Los nucleósidos y las bases se separarán mediante cromatografía líquida de fase reversa con una columna Symmetry C-18, de acuerdo a los métodos previamente descritos.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 43 de 82
6.5 Variable terciaria: Se estudiará el efecto del tratamiento con capecitabina sobre la aparición de amenorrea en mujeres premenopáusicas antes del tratamiento, y duración de la misma, mediante el cuestionario de la Dra. Cobleigh, del Hospital Rush Presbyterian St-Luke de Chicago (apéndice 7). Se definirá antes de la inclusión en el estudio el estado menopáusico de cada paciente, en función de las definiciones siguientes: a) Premenopáusica ?? No histerectomía previa; menstruación en los 6 meses previos a la randomización. ?? Histerectomía previa; y ó < 40 años de edad, ó > 40 años de edad con valores de LH y FSH
premenopáusicos. b) Postmenopáusica/Perimenopáusica ?? No histerectomía previa ; AUSENCIA de menstruación en los 6 meses previos a la
randomización. Histerectomía previa; y ó > 55 años de edad, ó < 55 años de edad con valores de LH y FSH postmenopáusicos. Se administrarán los cuestionarios de estado menstrual a las pacientes que se clasifiquen como premenopáusicas en el formulario de randomización.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 44 de 82
7. EVALUACIONES DEL ESTUDIO Se llevarán a cabo procedimientos diagnósticos completos para determinar el estadio en los meses previos a la inclusión en el estudio. Si se dispone del estudio de extensión previo al comienzo de la quimioterapia estándar, no será necesario repetir las pruebas. Todas las pacientes deberán disponer de una mamografía bilateral, radiografía de tórax (PA y lateral), ecografía abdominal y/o TAC toraco-abdominal. En caso de dolores óseos y/o elevación de las cifras de fosfatasa alcalina deberá realizarse, además gammagrafía ósea: no obstante, se recomienda realizar una gammagrafía ósea basal en todos los pacientes incluidos en el estudio. Se pueden realizar otras pruebas, según esté indicado clínicamente. 7.1 Calendario de pruebas (x: obligatorias; ? : si clínicamente indicado; rd: randomización; QT: quimioterapia; Brazo A: capecitabina; Brazo B: observación) 0= día de la rd Antes
de la rd Brazo A Brazo B
Día del ciclo Durante QT
Años 1-2
Años 3-5 Años 1-2 Años 3-5
Frecuencia Día 1 cada ciclo
Cada 3 meses
Cada 6 meses
Cada 3 meses
Cada 6 meses
Consentimiento informado
X
Historia X X X X X X Examen físico X X X X Análisis en sangre Hematología X X ? ? Bioquímica X ? ? Farmacogenética** X Análisis en sangre u orina
Prueba de embarazo X Evaluación del tumor Mamografía X ? ? Radiografía tórax X ? ? Ecografía o TAC abdominal
X ? ?
Gammagrafía ósea ? Evaluaciones en tejido tumoral Estado receptores hormonales*
X
Estado HER2* X Otros Acontecimientos adversos
X X X X X
Evaluación amenorrea (sólo en mujeres premenopáusicas)
X X X X X X
Estado menopáusico X X X X X * A realizar en el laboratorio central designado. ** Sólo en pacientes seleccionadas.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 45 de 82
Las evaluaciones de laboratorio se realizarán, siempre que sea posible, en el mismo laboratorio durante todo el estudio. Se procurará utilizar la misma exploración radiológica desde la evaluación basal hasta el seguimiento. 7.2 Criterios de retirada y análisis previstos de retiradas y abandonos Los pacientes podrán retirarse del estudio en los casos siguientes:
a. Ante recidiva de la enfermedad, aparición de una segunda neoplasia primaria (con excepción del cáncer de piel no melanoma tratado con intención curativa, o el carcinoma in situ de cérvix), muerte o administración de otro tratamiento antineoplásico sistémico distinto del fármaco en estudio.
b. Toxicidad inaceptable a criterio del investigador. c. Interrupción de la dosis de capecitabina durante más de 3 semanas. d. Enfermedad intercurrente u otros motivos que, en opinión del investigador, afecten
significativamente la valoración del estado clínico y que requieran la interrupción del tratamiento.
e. Petición de retirada por parte del paciente. f. Decisión del promotor (por ejemplo, por violación del protocolo).
El motivo y la fecha de la suspensión del tratamiento se documentará, para todas las pacientes, en el cuaderno de recogida de datos (Ej. finalización del estudio, acontecimiento adverso, pérdida para el seguimiento). Se hará el seguimiento regular de las pacientes que salgan del estudio por cualquier motivo que no sea el de recibir una terapia antineoplásica sistémica por la recaída de la enfermedad o por una segunda neoplasia primaria. 7.3 Terapia después de suspender el tratamiento del protocolo Si las pacientes salen del estudio debido a progresión de la enfermedad, la administración de un tratamiento adicional dependerá del criterio del investigador. No se permite administrar otra terapia antitumoral (cirugía, quimioterapia, inmunoterapia), antes de que se documente la progresión tumoral. Si esto no es posible, se considerará que la paciente ha manifestado progresión en la fecha en que se inicie la nueva terapia antitumoral y se analizará en la categoría de fracaso a efectos del cálculo de la supervivencia libre de enfermedad. 7.4 Acontecimientos adversos
Definición de un acontecimiento adverso El investigador instruirá a las pacientes para que comuniquen la aparición de cualquier acontecimiento adverso. Un acontecimiento adverso es cualquier efecto no deseable por su intensidad y/o frecuencia, que puede estar asociado con el uso de un fármaco, se considere o no relacionado con el mismo, e incluye cualquier efecto secundario, lesión, toxicidad o reacciones de sensibilidad. También incluye cualquier cambio clínico o de laboratorio no deseable que normalmente no se manifestaría en el paciente. ?? Acontecimiento adverso grave (AAG) Un acontecimiento adverso grave es aquel que sugiere un riesgo significante, una contraindicación, un efecto secundario o una precaución. Se considera acontecimiento adverso grave, aquel que, a cualquier dosis, cumple al menos uno de los siguientes criterios:
?? Provoca la muerte (si la misma no se encuadra en la definición de efecto de eficacia).

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 46 de 82
?? Amenaza la vida (significa que el paciente corría el riesgo inmediato de fallecer a causa del acontecimiento en el momento de producirse. No incluye un acontecimiento que, de haberse manifestado de forma más grave, podría haber provocado la muerte.)
?? Requiere o prolonga la hospitalización del paciente (la hospitalización se refiere a una ingreso hospitalario no programado).
?? Da lugar a incapacidad o discapacidad persistente o significativa ?? Provoca una anomalía congénita o un defecto de nacimiento ?? Se considera un acontecimiento clínicamente importante o requiere una intervención
para prevenir cualquiera de los desenlaces anteriores). Los acontecimientos clínicamente importantes son aquellos que pueden no ser inmediatamente amenazantes para la vida, pero claramente tienen una significación clínica relevante. Pueden poner en peligro al sujeto o requerir una intervención para evitar cualquier resultado grave. El cáncer o la sobredosificación o el abuso de un fármaco se considerarán, normalmente, como graves. Se deberá ejercer el juicio médico y científico a la hora de decidir la notificación al promotor en otras situaciones, tales como eventos médicos que pueden no ser amenazantes para la vida de inmediato, o provocan la muerte o la hospitalización, pero que ponen en riesgo al paciente o pueden requerir una intervención para prevenir un desenlace de la lista anterior. Estas situaciones también se consideran generalmente como acontecimientos adversos graves. Ejemplos de tales acontecimientos son la asistencia al servicio de urgencias o la asistencia domiciliaria, debido a broncoespasmo, discrasias de la sangre o convulsiones que no provocan hospitalización; o desarrollo de drogodependencia o abuso de drogas. Un acontecimiento adverso inesperado es el que por su naturaleza o severidad no aparece reflejado en la ficha técnica del producto. El investigador debe evaluar inicialmente la relación causal. Existen dos posibilidades:
?? No (no relacionado con el fármaco en estudio). ?? Sí (relación remota, posible, probable o definitiva con el fármaco en estudio).
El término “severo” se usa como medida de intensidad, aunque un acontecimiento adverso severo no tiene necesariamente que ser grave. Por ejemplo, náuseas de varias horas de duración se pueden clasificar como severas, pero no son clínicamente graves.
Todos los acontecimientos adversos graves que se manifiesten durante el período de tratamiento del estudio o en los 30 días posteriores a la administración del fármaco del estudio, se deben comunicar según el procedimiento descrito a continuación. Para cualquier AAG tardío (que se manifieste después de este período de 30 días) que esté posible o probablemente relacionado con el tratamiento del estudio, se debe seguir el mismo procedimiento para su comunicación. La progresión de alguna enfermedad subyacente del paciente, que dé lugar a uno de los acontecimientos anteriores, se deberá comunicar como experiencia adversa grave (pero esperada) que (a) no está relacionada con la medicación del estudio o (b) está producida por el fracaso del efecto terapéutico anticipado del fármaco del estudio. Si el acontecimiento adverso es grave, se debe comunicar en el plazo de un día laborable , desde que el investigador tiene noticia del mismo, al coordinador administrativo del estudio, en el Departamento Central de Operaciones de CIBOMA, utilizando la hoja de notificación de acontecimientos adversos que se proporciona en el archivo del investigador:

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 47 de 82
CIBOMA Teléfono: +34 916592870
Fax: +34 916510406 La Sede Científica del CIBOMA notificará al responsable de farmacovigilancia de la compañía farmacéutica fabricantes del fármaco del estudio, a las autoridades sanitarias de los países participantes, a los Comités de Ensayos Clínicos (CEIC) y a los investigadores, en los plazos de tiempo establecidos por la normativa española vigente, todos los AAG e inesperados que puedan estar relacionados con los tratamientos en investigación que hayan ocurrido en nuestro país. La terminación prematura del estudio y las medidas terapéuticas que se deben adoptar dependerán del criterio del investigador. Se proporcionará una explicación completa en el cuaderno de recogida de datos apropiado sobre la terminación prematura del estudio. El investigador realizará un seguimiento de todos los acontecimientos adversos, independientemente de su severidad, hasta su resolución satisfactoria. El investigador y las personas encargadas del cuidado del paciente deberán iniciar cualquier investigación complementaria de los acontecimientos adversos graves basándose en su opinión clínica sobre los posibles factores causantes. Este procedimiento puede incluir la necesidad de procurar la opinión adicional de un especialista del campo del acontecimiento adverso. Si alguna paciente fallece, se deberán proporcionar a CIBOMA todos los hallazgos post-mortem, incluida la histopatología. Las demás reacciones adversas se recopilarán en el cuaderno de recogida de datos (CRD) durante el estudio. De acuerdo con las disposiciones de este protocolo, el investigador deberá aceptar estas responsabilidades. Muertes durante el estudio Cualquier fallecimiento que sobrevenga durante la parte de tratamiento activo del estudio y en los 30 días posteriores a la última administración, se deberá comunicar a la Sede Científica de CIBOMA en el plazo de 24 horas, independientemente de su relación con el fármaco o fármacos del estudio. Transcurridos 30 días desde la última administración de un fármaco del estudio, sólo será necesario comunicar las muertes como acontecimientos adversos graves si se considera que tienen una posible relación con la medicación del estudio. Todas las muertes se deberán comunicar en la sección del CRD correspondiente al informe de fallecimiento, independientemente de su causa. Se deberá documentar la causa del fallecimiento (relacionado con el cáncer, relacionado con el tratamiento, no relacionado con el cáncer ni con el tratamiento).

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 48 de 82
8. RECOGIDA Y PROCESAMIENTO DE LOS DATOS DEL ESTUDIO 8.1 Monitorización, auditoria e inspección El estudio se monitorizará según normas de buena práctica clínica (BPC), mediante visitas regulares al centro y conversaciones telefónicas que el monitor del estudio mantendrá con el investigador. Durante las visitas al centro, el monitor deberá revisar los datos originales del paciente, los impresos de contabilización de la medicación y los archivos de documentación. Además, el monitor deberá observar los procedimientos del estudio y comentar cualquier problema con el investigador. En el transcurso del estudio, las Autoridades Sanitarias pueden realizar una visita de inspección al centro. El investigador proporcionará el acceso directo a los datos originales / documentos para la monitorización relacionada con el ensayo, las auditorias, la revisión por el Comité Ético de Investigación Clínica y las Autoridades Sanitarias. 8.2 Registro de los datos CIBOMA proporcionará los Cuadernos de Recogida de Datos (CRD), en formato electrónico, para que los investigadores puedan grabar la información de las pacientes del estudio en el mismo. CIBOMA dispone de un Servidor seguro (certificado SSL) dedicado en exclusiva a la grabación de los datos del cuaderno de recogida de datos electrónico. Se ha contratado un ancho de banda necesario para una buena conexión. CIBOMA garantiza el mantenimiento de dicho servidor, así como de la programación del cuaderno de recogida de datos, durante todo el estudio. El cuaderno de recogida de datos electrónico incluirá la firma electrónica para acceso autorizado, un módulo de programación de la trazabilidad de la información grabada, los cambios realizados, y el bloqueo automático tras el tiempo establecido desde la grabación. La grabación de datos en el CRD podrá ser directa (acceso en tiempo real desde el punto de origen) o diferida, para lo que se programará un sistema de replicación y transmisión a la conexión. El CRD se diseñará con una especificación de filtros desarrollada por el departamento de Gestión de Datos de CIBOMA. El propósito de los filtros es chequear las omisiones de datos, correcciones, discrepancias y clarificaciones. Los filtros son realizados para aumentar la precisión tanto de la entrada de datos como de los Cuadernos de Recogida de Datos. Después de la entrada y verificación de los datos, se ejecutará el programa de validación; tras lo cual se imprimirán las queries que serán enviadas al investigador. El monitor deberá verificar los CRD, cotejándolos con todos los datos originales (historia clínica, informes de especialistas médicos, etc). Al final del estudio, se proporcionarán copias impresas de todos los CRD de pacientes reclutadas en cada centro participante. El investigador principal deberá emitir un certificado de conformidad con los contenidos de cada CRD, que se guardará en el archivo central del estudio. El Cuaderno de Recogida de Datos para este estudio será de diseño electrónico cumpliendo con las normativas internacionales ISO para la seguridad de información en "redes informáticas" y otros medios por donde fluye información y las IEEE para la estandarización de los planes de verificación, validación y documentación de la programación utilizada para la creación del Cuaderno de Recogida de Datos Electrónico. A su vez se cumplirá la normativa de la FDA para los registros y firmas electrónicos, GMP y GLP, así como la normativa de los sistemas de calidad.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 49 de 82
8.3 Identificación de los datos que se pueden recoger directamente en el CRD y que serán considerados así documentos fuente. Se acepta que los siguientes datos se pueden transcribir directamente en el CRD: ?? Peso de la paciente antes de cada ciclo (excepto en la primera visita, en la que es necesario calcular la superficie corporal). ?? Estado funcional en cada visita (excepto en la previa a la randomización- criterio de inclusión).

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 50 de 82
9. CONSIDERACIONES ESTADÍSTICAS 9.1 Poblaciones a analizar El análisis de la eficacia primaria se realizará en la población de intención de tratar (ITT), que se define como la población de todas las pacientes randomizadas, analizadas en el grupo de tratamiento que se les ha asignado. Las pacientes randomizadas que no recibieron quimioterapia se analizarán en su grupo de randomización. El análisis ITT se realizará para la SLE y SG. Además, los análisis de supervivencia se realizarán en la población de pacientes seleccionadas , compuesta por aquellas pacientes que no hayan incurrido en ninguna violación mayor del protocolo. El análisis de la toxicidad y el de calidad de vida se realizará en todas las pacientes del estudio que hayan recibido al menos 1 ciclo de tratamiento, o que hayan completado el periodo de observación equivalente a 1 ciclo. 9.2 Métodos estadísticos Las variables aleatorias continuas se expresarán como medida de tendencia central: media y/o mediana y medida de dispersión: desviación típica y rango. Las variables de tipo cuantitativo se expresarán como porcentajes. En todos los casos que sea posible, se calculará el intervalo de confianza al 95% de cada una de las estimaciones realizadas. Se utilizará el método del límite-producto de Kaplan-Meier para estimar la SLE y la SG. La comparación de estos 2 parámetros entre los dos grupos de tratamiento se realizará utilizando la prueba Log Rank. Todos los tests de las hipótesis serán bilaterales. Los intervalos de confianza al 95% de las medianas de supervivencia se calcularán utilizando el método de Simon. Además, se realizará un análisis de regresión múltiple de Cox para la SLE y SG, con el fin de ajustar la comparación del tratamiento para los factores pronósticos principales. Estos factores incluyen la edad, el estado menopáusico, el tipo de cirugía, los hallazgos histopatológicos, y el tamaño del tumor. Se utilizará la prueba de Wald para establecer la importancia pronóstico de cada covariable. Las covariables que parezcan no balanceadas en los valores basale s se añadirán posiblemente al modelo de Cox. El análisis por subgrupos se realizará únicamente si el grado de significación estadística en la prueba preliminar es muy alto (p<0,01). En el análisis estadístico, un centro corresponderá a un hospital participante. Se prevé contar al final del estudio con un gran número de centros con pocos pacientes por cada uno de ellos. Por consiguiente, no se ha planeado incluir ningún efecto del centro en el modelo estadístico. Sin embargo, en caso de que en algunos centros se produzca una diferencia considerable en el reclutamiento, se ha planeado comparar la consistencia de los resultados entre estos centros y los resultados de todo el estudio, en términos de las características basales principales y el objetivo primario. 9.3 Evaluación de la seguridad Para calificar los acontecimientos adversos para su registro en el CRD, se utilizarán los Criterios de Toxicidad Común del Instituto Nacional del Cáncer (NCI-CTC) y el sistema de clasificación correspondiente. Para todos los acontecimientos adversos que no se clasifiquen en el NCI-CTC, se aplicará la clasificación de COSTART (FDA 1989) (severidad = 1: leve, 2: moderada 3: grave y 4: amenazante para la vida).

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 51 de 82
Los acontecimientos adversos se compararán utilizando el test bilateral Cochran Mantel Haenszel, el test de la ?2 de Pearson o el test exacto de Fisher, bilaterales. En vista del gran número de test estadísticos previstos, los valores p no se interpretarán en el sentido habitual, sino que se utilizarán como un “instrumento señalizador” para destacar las diferencias que justifiquen una atención adicional
9.4 Determinación del tamaño de la muestra Los siguientes datos se han obtenido a partir de la información de la base de datos del proyecto “El Álamo”. En total se han considerado 1627 pacientes entre los años 1990 y 1997. La población está formada por pacientes con cáncer de mama operable, cirugía, ganglios positivos y receptores hormonales negativos, o ganglios negativos, receptores hormonales negativos y tumores T2-3. Para estos grupos de pacientes, la supervivencia libre de enfermedad estimada a los 5 años es del 64,72%. Se pretende detectar una reducción del 25% en la razón de riesgos, con una potencia del 80% usando la prueba log-rank a un nivel bilateral del 0,05. En este caso, la SLE a 5 años en el brazo experimental sería del 72,2%. Esto requiere que el número de eventos para la SLE sea de 379 en total. SLE a 5 años en el brazo control = 64,72%
Prueba Log-rank Alfa = 0.05 bilateral; potencia = 80%. Diferencia
relativa Hazard ratio Nº eventos Pacientes por rama Total
25% 0,75 379 601 1203
Considerando un 10% de pérdidas post-randomización, el tamaño final será de 1324 pacientes, 662 por brazo de tratamiento. 9.5 Análisis estadístico de los datos de farmacogenética de TS y MTHFR 9.5.1 Hipótesis: Se estima que se detectará una diferencia del 40% en la toxicidad global grado ¾ entre los pacientes 2R/2R y los 3R/3R, siendo estos últimos los que presenten menor toxicidad. Se espera que los homocigotos TT tendrán mayor toxicidad frente a la capecitabina y que se hallará significación clínica mediante estratificación por polimorfismos en la zona del promotor de TS. 9.5.2 Cálculo del tamaño de muestra: Los requerimientos de tamaño de muestra se basan en la variable principal del estudio, no en las variables farmacogenéticas. Sin embargo, el estudio dispondrá de potencia estadística para determinar si los polimorfismos en el promotor de TS se asocian a diferencias en la tasa de toxicidad agregada. En un estudio retrospectivo en pacientes con cáncer de colon metastásico, las tasas de toxicidad del 63% y el 27% se asociaron a los polimorfismos 2R/2R y 3R/3R respectivamente53. Los alelos más frecuentes en caucasianos y asiáticos son los 2R y 3R. Por el contrario, los alelos 4R, 5R y 9R son más frecuentes en descendientes de africanos52. Las frecuencias de alelos de repetición en tandem 3R y 2R esperadas son 0,6 y 0,4 respectivamente, y las frecuencias genotípicas asumidas de 2R/2R, 2R/3R y 3R/3R son 0,16, 0,48 y 0,36, respectivamente. Para detectar una diferencia en la toxicidad agregada entre 2R/2R (60%) y 3R/3R (20%), usando el test chi cuadrado bilateral con alfa=0,05 y beta= 0,20, se requerirán un mínimo de 17 pacientes 2R/2R y 37 pacientes 3R/3R. La frecuencia de homocigóticos MTHFR en la población caucásica es de al menos un 10%64. El estudio de Cohen et al63 permite estimar un tamaño de muestra necesario para estudiar los

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 52 de 82
efectos de MTHFR sobre la eficacia de la capecitabina. Se observaron tasas de respuesta del 58% en los individuos con genotipo CC y CT comparado con la tasa de respuesta del 100% en individuos con genotipo TT. Estas estimaciones de respuesta y frecuencia alélica sugieren que se precisará un tamaño de muestra de 80 pacientes como mínimo, para un alfa =0,05 y un beta= 0,8. 9.5.3 Análisis estadístico: Las pacientes en que se encuentren deficiencias en DPD serán excluidas del análisis para evitar interferencias en la interpretación de los datos de genotipos TS y toxicidad. Las pacientes que presenten toxicidad durante el tratamiento con capecitabina se agruparán en: i) grado ½; ii) grado 3/4,. El análisis primario comparará las tasas de toxicidad global grado 3 /4 entre los tres genotipos TS usando la prueba chi cuadrado, seguido de un análisis descriptivo de las toxicidades específicas (diarrea, mucositis, neutropenia y eritrodisestesia palmo-plantar). Se realizarán modelos de asociación de toxicidades y genotipos de TS y MTHFR usando pruebas de regresión logística. Con respecto al análisis de supervivencia, se calcularán las curvas de Kaplan-Meier para cada genotipo TS, y se compararán con las curvas generadas por el test Log Rank, con niveles de significación establecidos en un p<0,05 en análisis bilaterales. No están planeados ajustes por análisis múltiples. Se realizarán las siguientes comparaciones: i) Sólo en las pacientes que reciben capecitabina, se comparará la supervivencia libre de enfermedad entre los individuos con genotipo 2R/2R y los que presenten genotipos diferentes. ii) En pacientes que no reciben capecitabina, se comparará la supervivencia libre de enfermedad entre pacientes con genotipo 2R/2R y los que presentan genotipos diferentes, para descartar que el genotipo sea un factor pronóstico. iii) Se comparará la supervivencia libre de enfermedad entre los individuos que presentan genotipo 2R/2R tratados con capecitabina, y los del mismo genotipo no tratados con capecitabina. iv) Se comparará la supervivencia libre de enfermedad entre los individuos que presentan genotipo 2R/3R tratados con capecitabina, y los del mismo genotipo no tratados con capecitabina. v) Se comparará la supervivencia libre de enfermedad entre los individuos que presentan genotipo 3R/3R tratados con capecitabina, y los del mismo genotipo no tratados con capecitabina.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 53 de 82
8 ASPECTOS PRÁCTICOS 10.1 Comité Independiente de Evaluación de Datos (IDMC) 10.1.1 Composición y misión del IDMC
El Comité Independiente de Evaluación de Datos (IDMC) estará compuesto por tres médicos oncólogos y un especialista en estadística. Estos miembros serán independientes del estudio clínico y estarán familiarizados con la metodología de los estudios oncológicos. Deberán ser conscientes de los peligros de tomar conclusiones sobre la base de datos inmaduros y estarán de acuerdo con el diseño y los objetivos de este protocolo. La misión del IDMC será la realización ética del estudio clínico y la protección de los intereses en cuanto a la seguridad de las pacientes que participen en el estudio. Este comité asegura la factibilidad y progreso del estudio clínico. El IDMC será responsable tanto de la revisión de la eficacia del estudio como de su seguridad. 10.1.2 Reuniones del IDMC Se convocará a una reunión anual del IDMC en ausencia de cualquier circunstancia importante que requiera de la reunión de sus miembros. El comité se reunirá anualmente para evaluar la incidencia y severidad de cada uno de los acontecimientos graves o no graves. En el caso en que se notifique un acontecimiento grave no anticipado o incidente antes de una reunión programada, se convocará una reunión inmediatamente para evaluar y asegurar la seguridad de los pacientes en este estudio. Esto último podría incluir también datos procedentes de otros estudios pero que implican los mismos agentes que están siendo administrados en este estudio. Esto último podría también incluir nuevos datos presentados sobre eficacia de otros estudios relevantes, cuyos resultados podrían, de alguna manera, influir sobre el estudio CIBOMA/2004-01. Los procedimientos y operaciones del IDMC se registrarán por escrito, levantándose acta de todas sus reuniones. 10.1.3 Recomendaciones del IDMC Al finalizar cada reunión del IDMC se recomendará por escrito la modificación del estudio (incluyendo las razones) o hacer públicos los resultados del análisis intermedio (incluyendo las razones) o continuar con el estudio sin modificaciones. 10.2 Presupuesto del estudio Se dispone de una partida presupuestaria asignada a este estudio para honorarios de los investigadores. Se abonarán a los hospitales participantes los costes indirectos generados por la realización del estudio. No se estima que existan costes directos extraordinarios, ya que las pruebas diagnósticas son estándar para este tipo de paciente. El presupuesto detallado se entrega en documento aparte. 10.3 Seguro de responsabilidad civil CIBOMA ha contratado un seguro de responsabilidad civil con la compañía Gerling-Konzern, que cubre a todos los sujetos participantes en el estudio de los posibles perjuicios que pudiera sufrir el paciente como consecuencia de su participación en el ensayo. Se enviará a los Comités Éticos de Investigación Clínica involucrados un certificado de la póliza de seguros del estudio específico para el mismo.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 54 de 82
10.4 Archivo de documentación El investigador deberá conservar las reproducciones de toda la información pertinente durante un período de, al menos, 15 años, a partir de la terminación del estudio. CIBOMA proporcionará al investigador un archivador para todos los documentos necesarios del estudio; asimismo, CIBOMA facilitará una lista de toda la documentación que se debe conservar. 10.5 Condiciones de publicación CIBOMA se compromete a publicar los resultados del estudio y a respetar los derechos de investigadores y centros participantes en el estudio.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 55 de 82
11. ASPECTOS ÉTICOS
11.1 Declaración de Helsinki Este estudio se llevará a cabo de acuerdo con la Declaración de Helsinki (enmienda de Edimburgo, 2000), descrita en el apéndice 1, siguiendo las Normas de Buena Práctica Clínica de la Conferencia Internacional de Armonización (GCP/ICH), y cumpliendo la legislación vigente en cada país participante. 11.2 Consentimiento Informado (apéndice 4) La paciente firmará en primer lugar un consentimiento informado que autorice el envío de su muestra tumoral al laboratorio central designado. Antes de la evaluación de selección, se informará a la paciente de la naturaleza de la medicación del estudio y se le facilitará información pertinente en cuanto a la finalidad perseguida, los posibles beneficios y las posibles experiencias adversas. Se explicarán los procedimientos y los posibles riesgos a los que se expondrá la paciente. A continuación la paciente y el investigador leerán y firmarán el consentimiento informado del estudio. Se facilitará a la paciente una copia del consentimiento informado firmado y fechado. La paciente puede abandonar el estudio en cualquier momento, sin perjuicio del tratamiento médico que pueda recibir en el futuro. La verificación del consentimiento informado firmado se registrará en la historia clínica de la paciente del estudio. 11.3 Confidencialidad El investigador se responsabilizará de conservar la información necesaria sobre cada paciente (Ej. iniciales del nombre, dirección, número de teléfono, número de seguridad social e identidad en el estudio), de manera que las autoridades sanitarias o CIBOMA puedan tener acceso a dicha información, en el caso de que sea necesario, manteniendo confidencial el nombre del paciente. Esta información se deberá conservar de manera confidencial todo el tiempo que se estipule legalmente, según la legislación española . 11.4 Comité Ético de Investigación Clínica (CEIC) Un CEIC debidamente constituido revisará el protocolo final aprobado y el consentimiento informado. La decisión del CEIC relativa a la realización del estudio se entregará por escrito a CIBOMA y al investigador. CIBOMA y/o el investigador aceptarán presentar al CEIC los informes necesarios sobre el progreso del estudio, así como comunicar cualquier acontecimiento adverso grave, los problemas que sean amenazantes para la vida del paciente o muertes. El investigador también informará al CEIC de los casos de acontecimientos adversos graves (que le haya proporcionado CIBOMA) comunicados en otros estudios clínicos realizados con el fármaco del estudio. El investigador deberá informar al CEIC de la terminación del estudio. 11.5 Modificaciones del protocolo Será considerada una modificación relevante al protocolo cualquier modificación que pueda repercutir en la realización del estudio, los beneficios potenciales para el paciente o pueda afectar a la seguridad del paciente, incluidos los cambios en los objetivos del estudio, diseño del estudio, población de pacientes, tamaño de las muestras, procedimientos del estudio o aspectos administrativos significativos. Dicha enmienda será autorizada por CIBOMA y el investigador, y

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 56 de 82
aprobada por el CEIC y las autoridades sanitarias antes de su aplicación, de acuerdo con la legislación local. Las modificaciones no relevantes del protocolo consisten en correcciones menores y/o aclaraciones que no repercuten en la forma en que se va a llevar a cabo el estudio. Dichas modificaciones no relevantes serán autorizadas por CIBOMA y el investigador y se documentarán en un memorándum. Las modificaciones no relevantes se notificarán al CEIC y a las autoridades sanitarias según la legislación local. 11.6 Identificación de los pacientes En la visita inicial, se registrarán cronológicamente en el archivo del investigador las iniciales y la fecha de nacimiento de las pacientes seleccionadas para el estudio. En el caso de que se excluya alguna paciente de la participación en el estudio, se deberá documentar el motivo en el espacio proporcionado a tal fin. Se asignará un número de estudio a cada paciente en el momento del registro. El Número de Asignación del Paciente se deberá registrar en el Cuaderno de Recogida de Datos.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 57 de 82
28. BIBLIOGRAFÍA
1. Henderson IC: Chemotherapy for metastatic disease, in: Harris JR, Hellman S, Henderson IC, Kinne DW eds: Breast diseases. Philadelphia: J.B. Lippincott compagny, 2nd edition, 1991: pp. 604-665.
2. National Institutes of Health Consensus Development Conference Statement: Adjuvant Therapy for Breast Cancer, November 1-3, 2000. J Natl Cancer Inst 2001; 93 (13): 979-989.
3. Aapro MS. Adjuvant therapy of primary breast cancer: a review of key findings from the 7th International Conference, St. Gallen, February 2001. The Oncologist 2001; 6: 376-385.
4. Bonadonna G, Brusamolino E, Valagussa P et al. Combination chemotherapy as an adjuvant treatment in operable breast cancer. N Engl J Med 1976; 294: 405.
5. Fisher B, Brown AM, Dimitrov et al. Two months of doxorubicin-cyclophosphamide with and without interval reinduction therapy compared with 6 months of cyclophosphamide, methotrexate, and fluorouracil in positive-node breast cancer patients with tamoxifen-non-responsive tumors: results from NSABP B-15. J Clin Oncol 1990; 8: 1483.
6. Carpenter JT, Velez-Garcia E, Aron BS et al. Five-year results of a randomized comparison of cyclophosphamide, doxorubicin (Adriamycin) and fluorouracil (CAF) vs. cyclophosphamide, methotrexate and flurouracil (CMF) for node positive breast cancer: a Southeastern Cancer Study Group Study. Proc ASCO 1994; 13: 66.
7. Buzzoni R, Bonadonna G, Valagussa P et al. Adjuvant chemotherapy with doxorubicin plus cyclophosphamide, methotrexate, and fluorouracil in the treatment of resectable breast cancer with more than three positive axillary nodes. J Clin Oncol 1991; 9: 2134.
8. Coombes RC, Bliss JM, Marty M et al. A randomized trial comparing adjuvant FEC with CMF in premenopausal patients with positive resectable breast cancer. Proc AmSoc Clin Onc 1991; 10 : 41.
9. Misset JL, Gil-Dalgado M, Chollet Ph et al. Ten-year results of the French trial comparing Adriamycin, vincristine, 5-fluorouracil and cyclophosphamide to standard CMF as adjuvant therapy for node positive breast cancer. Proc Am Soc Clin Onc 1992; 11 : 54.
10. Goldhirsch A, Glick J, Gelber R et al. Meeting Highlights: International Consensus Panel on the Treatment of Primary Breast Cancer. J. Natl. Cancer Inst. 1998; 90(21): 1601-1608.
11. Early Breast Cancer Trialists' Collaborative Group. Polychemotherapy for early breast cancer: an overview of the randomized trials. The Lancet 1998; 352; 930: 942.
12. M. Martín, A. Rodríguez-Lescure, A. Ruiz, E. Alba, L. Calvo, M. Ruiz-Borrego, B. Munárriz, JM. López-Vega, C. A. Rodríguez, C. Crespo, M. Ramos, JM. Gracia -Marco, A. Lluch. Estudio fase III, multicéntrico, randomizado, comparando 6 ciclos (ci) de FEC con 4 ci de FEC seguidos de 8 administraciones semanales de paclitaxel (P) como tratamiento quimioterápico adyuvante de pacientes con cáncer de mama operado y afectación axilar (CMAA): primer análisis de seguridad del estudio GEICAM 9906. Comunicación oral #22. IX Congreso de SEOM (2003).
13. Henderson IC, Berry DA, Demetri GD, Cirricione CT, Goldstein LJ, Martino S et al. J Clin Oncol 2003; 21 (6): 976-983.
14. Citron ML, Berry DA, Cirrincione C, Hudis C, Winer EP, Gradishar WJ et al. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leucemia Group B Trial 9741. J Clin Oncol 2003; 21 (8): 1431-1439.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 58 de 82
15. VogelCL et al. The role of growth factor support following neutropenic events in early stage breast cancer (BC) patients treated with adjuvant docetaxel, doxorubicin, and cyclophosphamide (TAC): A sub-analysis of BCIRG 001. Proc. ASCO 2004; abstract 677.
16. Investigational Drug Brochure (Ro 09-1978), December 1997. 17. Barton G et al. Human platelet derived growth factor is homologous to E. coli
thymidine phosphorylase. Protein Sci 1992;1:688-690. 18. Sumizana T et al. Thymidine phosphorylas activity associated with platelet-derived
endothelial cell growth factor. J Biochem 1993;114:9-14. 19. Toi M et al. Expression of platelet-derived endothelial cell growth factor thymidine
phosphorylase in human breast cancer. Int J Cancer 1995;64:79-82. 20. Griffiths L et al. The influence of oxygen tension and pH on the expression of platelet-
derived endothelial cell growth factor/thymidine phosphorylase in human breast tumor cells grown in vitro and in vivo. Cancer Res 1997;57:570-572.
21. Moghaddam A et al.Thymidine phosphorylase is angiogenic and promotes tumor growth. Proc Natl Acad Sci 1995;92:998-1002.
22. Folkman J .What is the role of thymidine phosphorylase in tumor angiogenesis? J Natl Canc Inst 1996;88:1091-1092.
23. Ishikawa T, Fukase Y,Yamamoto T et al. Antitumor activities of a novel fluoropyrimidine, N 4-pentyloxycarbonyl-5’-deoxy-5fluorocytidine (capecitabine). Biol Pharm Bull 1998;21:713-717.
24. Ishikawa T, Sekiguchi F, Fukase Yet al. Positive correlation between the efficacy of capecitabine and doxifluridine and the ratio of thymidine phosphorylase to dihydropyrimidine dehydrogenase activities in tumors in human cancers xenografts. Cancer Res 1998;15:680-685.
25. Meropol NG, Budman Dr, Creaven P et al. A phase I study of continuous twice daily treatment with capecitabine in patients with advanced and/or metastatic solid tumors. Annals of Oncology 1996; 7 (Suppl. 1): 87; (abstract ? 298).
26. Mackean M, Planting A, Twelves C et al. Phase I and pharmacologic study of intermittent twice-daily oral therapy with capecitabine in patients with advanced and/or metastatic cancer. J Clin Oncol 1998;16: 2977-85.
27. Taguchi T., Ishitani K., Saitoh K et al .A Japanese phase I study of continuous twice daily treatment with capecitabine in patients with advanced and/or metastatic solid tumors. Annals of Oncology 1996; 7 (Suppl. 1): 87 (abstract ? 299).
28. Dirix LY, Bisset D, Van Oosterom AT et al. A phase I study of capecitabine in combination with oral leuvocorin in patients with advanced and/or metastatic solid cancer. Proc Am Soc Clin Oncol 1996;15, 496 (abstract ? 1821).
29. Van Cutsem E, Findlay M, Osterwalder et al. Capecitabine, an oral fluoropyrimidine carbamate with substantial activity in advanced colorrectal cancer: results of a randomized pahse II study. J Clin Oncol 2000;18:1337-1345
30. Villalona-Calero M.A., Weiss GR, Burris HA et al. Phase I and pharmacokinetic study of the oral fluoropyrimidine capecitabine in combination with paclitaxel in patients with advanced solid malignancies. J Clin Oncol, 1999; 17:1915-1925
31. Pronk LC, Vasey P, Sparreboom A et al.A phase I and pharmacokinetic study of the combination of capecitabine and docetaxel in patients with advanced solid tumours. Br J Cancer 2000 ;83:22-29
32. Twelves C, Budman DR, Creaven PJ et al. Pharmacokinetics (PK) and Pharmacodynamics (PD) of Capecitabine in two phase I studies. Proc Am Soc Clin Oncol 1996;15, 476 (abstract ? 1509).
33. Reigner B, Verweij J, Dirix L et al. Effect of food on the pharmacokinetics of capecitabine and its metabolites following oral administration in cancer patients. Clin Cancer Res 1998;4:941-948.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 59 de 82
34. Schueller J, Cassidy J, Reigner BG et al. Tumor selectivity of XELODATM in colorectal cancer patients. Proc Am Soc Clin Oncol 1997; 16: 227a (abstract ? 797).
35. Blum J, Jones S, Buzdar A, et al. Multicenter phase II study of capecitabine in paclitaxel-refractary metastatic breast cancer. J Clin Oncol, 1999;17: 485-93.
36. Thuss-Patience PC, von Minckwitz G, Lück et al. Capecitabine: a new standard in metastatic breast cancer recurring after anthracycline and taxane-containing chemotherapy? Results of a multicenter phase II trial. Proc Am Soc Clin Oncol 2001;20:66b
37. Jakob A, Schupp M, Knop s et al. A phase II study of capecitabine in patients, who relapsed after high dose chemotherapy followed by peripheral blood stem cell transplantation for metastatic breast cancer. Proc Am Soc Clin Oncol 2001;20:55b
38. Moiseenko VM, O’Reilly SM, Dalbot DC et al. A comparative randomized phase-II study of Xeloda (capecitabine) and paclitaxel in patients with breast cancer progressing after anthracycline antibiotics. Vopr Onkol 2000;46: 285-9
39. Kusama M, Sano M, Ikeda T et al. A phase II study of XelodaTM (capecitabine) in patients with advanced/metastatic breast carcinoma. The cooperative group of capecitabine for breast carcinoma. Proc Am Soc Clin Oncol 2001;20:44b
40. O’Shaughnessy J, Moiseyenko V, Bell D, et al. A randomized phase II study of XelodaTM (capecitabine) vs CMF as first line chemotherapy of breast cancer in women aged greater than or equal to 55 years . Proc Am Soc Clin Oncol 1998;16:103a
41. O'Shaughnessy JA.The evolving role of capecitabine in breast cancer. Clin Breast Cancer. 2003; 4 (Suppl 1): S20-5.
42. Crawford J, Dale D, Lyman G et al. Suboptimal doping in adjuvant breast cancer chemotherapy: evidence from a nationwide survey. Breast Cancer Res Treat 2000; 64: 66 (abstr. # 241).
43. Furroleau P et al: Eur J. Cancer 2004; 40: 536-542. 44. O´Shaughnessy J et al: J Clin Oncol 2002; 20: 2812-2823. 45. The Oncology Report. Fall 2004. 46. Fraile RJ, Baker LH, Buroker TR, et al: Pharmacokinetics of 5-fluorouracil
administered orally, by rapid intravenous and by slow infusion. Cancer Res 40:2223-8, 1980
47. Naguib FN, el Kouni MH, Cha S: Enzymes of uracil catabolism in normal and neoplastic human tissues. Cancer Res 45:5405-12, 1985
48. Baccanari DP, Davis ST, Knick VC, et al: 5-Ethynyluracil (776C85): a potent modulator of the pharmacokinetics and antitumor efficacy of 5-fluorouracil. Proc Natl Acad Sci U S A 90:11064-8, 1993
49. Lamont EB, Schilsky RL: The oral fluoropyrimidines in cancer chemotherapy. Clin Cancer Res 5:2289-96, 1999
50. Reigner B, Blesch K, Weidekamm E: Clinical pharmacokinetics of capecitabine. Clin Pharmacokinet 40:85-104, 2001
51. Takeishi K, Kaneda S, Ayusawa D, et al: Nucleotide sequence of a functional cDNA for human thymidylate synthase. Nucleic Acids Res 13:2035-43, 1985
52. Marsh S, Ameyaw MM, Githang'a J, et al: Novel thymidylate synthase enhancer region alleles in African populations. Hum Mutat 16:528, 2000
53. Horie N, Aiba H, Oguro K, et al: Functional analysis and DNA polymorphism of the tandemly repeated sequences in the 5'-terminal regulatory region of the human gene for thymidylate synthase. Cell Struct Funct 20:191-7, 1995
54. Kawakami K, Omura K, Kanehira E, et al: Polymorphic Tandem Repeats in the Thymidylate Synthase Gene is Associated with Its Protein Expression in Human Gastrointestinal Cancers. Proceedings of The American Society of Clinical Oncology 17:1128, 1998

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 60 de 82
55. Kawakami K, Salonga D, Park JM, et al: Different lengths of a polymorphic repeat sequence in the thymidylate synthase gene affect translational efficiency but not its gene expression. Clin Cancer Res 7:4096-101, 2001
56. Park D, Soehlmacher J, Wu Z, et al: Human Thymidylate Synthase Gene Polymorphism Determines Response to Capecitabine Chemotherapy in Advanced Colorectal Cancer. Proceedings of The American Society of Clinical Oncology 20:514, 2001
57. Mandola MV, Stoehlmacher J, Muller-Weeks S, et al: A novel single nucleotide polymorphism within the 5' tandem repeat polymorphism of the thymidylate synthase gene abolishes USF-1 binding and alters transcriptional activity. Cancer Res 63:2898-904, 2003
58. Marsh S, McKay JA, Cassidy J, et al: Polymorphism in the thymidylate synthase promoter enhancer region in colorectal cancer. Int J Oncol 19:383-6, 2001
59. Pullarkat ST, Stoehlmacher J, Ghaderi V, et al: Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J 1:65-70, 2001
60. Kang SS, Wong PW, Zhou JM, et al: Thermolabile methylenetetrahydrofolate reductase in patients with coronary artery disease. Metabolism 37:611-3, 1988
61. Weisberg I, Tran P, Christensen B, et al: A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol Genet Metab 64:169-72, 1998
62. Kawakami K, Omura K, Kanehira E, et al: Methylenetetrahydrofolate reductase polymorphism is associated with folate pool in gastrointestinal cancer tissue. Anticancer Res 21:285-9, 2001
63. Cohen V, Panet-Raymond V, Sabbaghian N, et al: Methylenetetrahydrofolate Reductase Polymorphism in Advanced Colorectal Cancer: A Novel Genomic Predictor of Clinical Response to Fluoropyrimidine-based Chemotherapy. Clin Cancer Res 9:1611-5, 2003
64. Botto LD, Yang Q: 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am J Epidemiol 151:862-77, 2000
65. Ma J, Stampfer MJ, Giovannucci E, et al: Methylenetetrahydrofolate reductase polymorphism, dietary interactions, and risk of colorectal cancer. Cancer Res 57:1098-102, 1997
66. Weisberg IS, Jacques PF, Selhub J, et al: The 1298A-->C polymorphism in methylenetetrahydrofolate reductase (MTHFR): in vitro expression and association with homocysteine. Atherosclerosis 156:409-15, 2001
67. Christensen B, Arbour L, Tran P, et al: Genetic polymorphisms in methylenetetrahydrofolate reductase and methionine synthase, folate levels in red blood cells, and risk of neural tube defects. Am J Med Genet 84:151-7, 1999
68. Diasio RB, Beavers TL, Carpenter JT: Familial deficiency of dihydropyrimidine dehydrogenase. Biochemical basis for familial pyrimidinemia and severe 5-fluorouracil-induced toxicity. J Clin Invest 81:47-51, 1988
69. Tuchman M, Stoeckeler JS, Kiang DT, et al: Familial pyrimidinemia and pyrimidinuria associated with severe fluorouracil toxicity. N Engl J Med 313:245-9, 1985
70. Etienne MC, Lagrange JL, Dassonville O, et al: Population study of dihydropyrimidine dehydrogenase in cancer patients. J Clin Oncol 12:2248-53, 1994.
71. Rouzier R, Anderson K, Hess KR, et al. Basal and luminal types of breast cancer defined by gne expression patterns respond differently to neoadjuvant chemotherapy. San Antonio Breast Cancer Symposium, San Antonio, TX, 2004.
72. Carey LA, Dees EC, Sawyer L, et al. The triple negative paradox: primary tumour chemosensitivity of the basal-like breast cancer (BBC) phenotype. San Antonio Breast Cancer Symposium, San Antonio, TX, 2004.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 61 de 82
73. Vukelja SJ, Lombardo FA, James WD et al. Pyridoxine for palmar-plantar erythrodysesthesia syndrome. Annals of Int Med 1989;8:688-689.
74. Vukelja SL, Baker WJ, Burris HA et al. Pyridoxine therapy for palmar-plantar erythrodysesthesia syndrome associated with taxotere. J Natl Cancer Inst 1993;85:1432-1433.
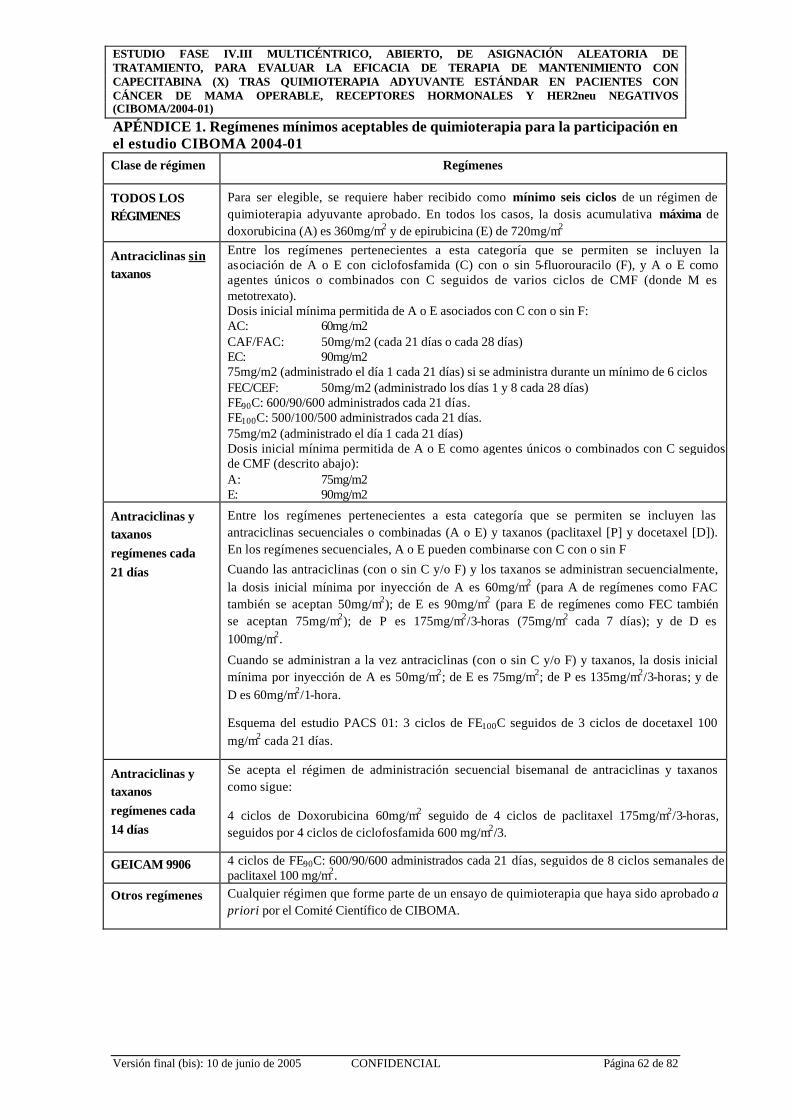
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 62 de 82
APÉNDICE 1. Regímenes mínimos aceptables de quimioterapia para la participación en el estudio CIBOMA 2004-01 Clase de régimen Regímenes
TODOS LOS RÉGIMENES
Para ser elegible, se requiere haber recibido como mínimo seis ciclos de un régimen de quimioterapia adyuvante aprobado. En todos los casos, la dosis acumulativa máxima de doxorubicina (A) es 360mg/m2 y de epirubicina (E) de 720mg/m2
Antraciclinas sin taxanos
Entre los regímenes pertenecientes a esta categoría que se permiten se incluyen la asociación de A o E con ciclofosfamida (C) con o sin 5-fluorouracilo (F), y A o E como agentes únicos o combinados con C seguidos de varios ciclos de CMF (donde M es metotrexato). Dosis inicial mínima permitida de A o E asociados con C con o sin F: AC: 60mg/m2 CAF/FAC: 50mg/m2 (cada 21 días o cada 28 días) EC: 90mg/m2 75mg/m2 (administrado el día 1 cada 21 días) si se administra durante un mínimo de 6 ciclos FEC/CEF: 50mg/m2 (administrado los días 1 y 8 cada 28 días) FE90C: 600/90/600 administrados cada 21 días. FE100C: 500/100/500 administrados cada 21 días. 75mg/m2 (administrado el día 1 cada 21 días) Dosis inicial mínima permitida de A o E como agentes únicos o combinados con C seguidos de CMF (descrito abajo): A: 75mg/m2 E: 90mg/m2
Antraciclinas y taxanos regímenes cada 21 días
Entre los regímenes pertenecientes a esta categoría que se permiten se incluyen las antraciclinas secuenciales o combinadas (A o E) y taxanos (paclitaxel [P] y docetaxel [D]). En los regímenes secuenciales, A o E pueden combinarse con C con o sin F
Cuando las antraciclinas (con o sin C y/o F) y los taxanos se administran secuencialmente, la dosis inicial mínima por inyección de A es 60mg/m2 (para A de regímenes como FAC también se aceptan 50mg/m2); de E es 90mg/m2 (para E de regímenes como FEC también se aceptan 75mg/m2); de P es 175mg/m2/3-horas (75mg/m2 cada 7 días); y de D es 100mg/m2.
Cuando se administran a la vez antraciclinas (con o sin C y/o F) y taxanos, la dosis inicial mínima por inyección de A es 50mg/m2; de E es 75mg/m2; de P es 135mg/m2/3-horas; y de D es 60mg/m2/1-hora.
Esquema del estudio PACS 01: 3 ciclos de FE100C seguidos de 3 ciclos de docetaxel 100 mg/m2 cada 21 días.
Antraciclinas y taxanos regímenes cada 14 días
Se acepta el régimen de administración secuencial bisemanal de antraciclinas y taxanos como sigue:
4 ciclos de Doxorubicina 60mg/m2 seguido de 4 ciclos de paclitaxel 175mg/m2/3-horas, seguidos por 4 ciclos de ciclofosfamida 600 mg/m2/3.
GEICAM 9906 4 ciclos de FE90C: 600/90/600 administrados cada 21 días, seguidos de 8 ciclos semanales de paclitaxel 100 mg/m2.
Otros regímenes Cualquier régimen que forme parte de un ensayo de quimioterapia que haya sido aprobado a priori por el Comité Científico de CIBOMA.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 63 de 82
APÉNDICE 2: ÍNDICE DEL ESTADO FUNCIONAL DE KARNOFSKY Y CRITERIOS DEL EASTERN COOPERATIVE ONCOLOGY GROUP (ECOG) ECOG KARNOFSKY DEFINITIONS (Zubrod) 0 100 Ausencia de síntomas.
1 80-90 Sintomático, completamente ambulatorio. 2 60-70 Sintomático, en cama menos de 50% del
día.
3 40-50 Sintomático, en cama más del 50% del día, pero no encamado.
4 20-30 Encamado.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 64 de 82
APÉNDICE 3: TNM DE MAMA (American Joint Committee on Cancer 2002) TUMOR PRIMARIO (T): TX No Se puede evaluar el tumor primario T0 Sin evidencia del tumor primario Tis Carcinoma in situ Tis (DCIS) carcinoma ductal in situ Tis (LCIS) carcinoma lobular in situ Tis enfermedad de Paget del pezón sin tumoración (si hay tumor asociado, se clasifica de
acuerdo a el tamaño de ese tumor). T1 Tumor de hasta 2 cm de diámetro. T1mic microinvasión de 0,1 cm o inferioe en su dimensión mayor T1a Hasta 0,5 cm de diámetro T1b Más de 0,5 cm y menos de 1 cm de diámetro T1c Más de 1 cm de diámetro y menos de 2 cm. T2 Tumor de más de 2 cm y menos de 5 cm en su diámetro mayor. T3 Tumor de más de 5 cm en alguno de sus diámetros T4 Tumor que, independientemente de su tamaño, infiltra directamente en la pared
torácica o piel. T4a Extensión hasta la pared torácica (que incluye costillas, músculos intercostales y
serrato anterior, pero no el músculo pectoral). T4b Edema (también piel de naranja) o ulceración de la piel de la mama, o nódulos
cutáneos salientes dentro de la misma mama. T4c Presencia de T4a más T4b. T4d Carcinoma inflamatorio. GANGLIOS LINFÁTICOS (pN) pNX No se pueden evaluar los ganglios linfáticos regionales (porque se hayan extirpado
anteriormente, o no se hayan extirpado) pN0 Sin presencia histológica de metástasis en ganglios linfáticos regionales, no se realiza un
exámen adicional para búsqueda de células tumorales aisladas . pN0(i-) Sin presencia histológica de metástasis en ganglios linfáticos regionales, inmunohistoquímica
negativa. pN0(I+) Sin presencia histológica de metástasis en ganglios linfáticos regionales, inmunohistoquímica
positiva en ausencia de agregados celulares mayores de 0,2 mm. pN0(mol-) Sin presencia histológica de metástasis en ganglios linfáticos regionales, sin hallazgos
moleculares por técnica de reacción de polimerasa en cadena-transcriptasa reversa (RT-PCR). pN0(mol+) Sin presencia histológica de metástasis en ganglios linfáticos regionales, hallazgos
moleculares positivos por técnica de reacción de polimerasa en cadena-transcriptasa reversa (RT-PCR).
pN1 Metástasis en 1 a 3 ganglios linfáticos axilares y/o cadena mamaria interna con enfermedad microscópica detectada por disección de ganglio centinela, pero no clínicamente aparente*.
pN1mi Micrometástasis (mayor de 0,2 mm y menor de 2 mm). pN1a Metástasis en 1 a 3 ganglios linfáticos axilares. pN1b Metástasis en cadena mamaria interna con enfermedad microscópica detectada por disección
de ganglio centinela pero no clínicamente aparente*. pN1c Metástasis en 1 a 3 ganglios axilares linfáticos y en cadena mamaria interna con enfermedad
microscópica detectada por disección de ganglio centinela pero no clínicamente aparente*. pN2 Metástasis en entre 4 y 9 ganglios axilares linfáticos o en ganglios mamarios internos
clínicamente aparentes* en ausencia de afectación ganglionar axilar. pN2a Metástasis en entre 4 y 9 ganglios axilares linfáticos (al menos un depósito tumoral mayor de
2 mm).
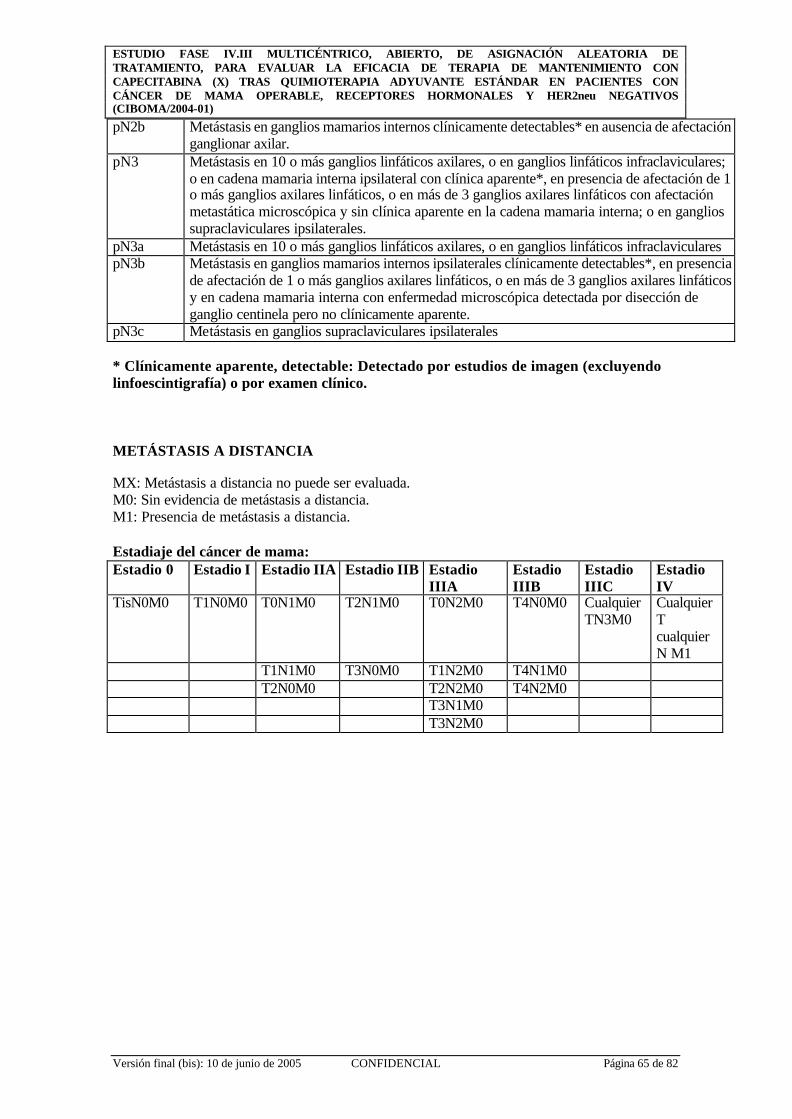
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Página 65 de 82
pN2b Metástasis en ganglios mamarios internos clínicamente detectables* en ausencia de afectación ganglionar axilar.
pN3 Metástasis en 10 o más ganglios linfáticos axilares, o en ganglios linfáticos infraclaviculares; o en cadena mamaria interna ipsilateral con clínica aparente*, en presencia de afectación de 1 o más ganglios axilares linfáticos, o en más de 3 ganglios axilares linfáticos con afectación metastática microscópica y sin clínica aparente en la cadena mamaria interna; o en ganglios supraclaviculares ipsilaterales.
pN3a Metástasis en 10 o más ganglios linfáticos axilares, o en ganglios linfáticos infraclaviculares pN3b Metástasis en ganglios mamarios internos ipsilaterales clínicamente detectables*, en presencia
de afectación de 1 o más ganglios axilares linfáticos, o en más de 3 ganglios axilares linfáticos y en cadena mamaria interna con enfermedad microscópica detectada por disección de ganglio centinela pero no clínicamente aparente.
pN3c Metástasis en ganglios supraclaviculares ipsilaterales * Clínicamente aparente, detectable: Detectado por estudios de imagen (excluyendo linfoescintigrafía) o por examen clínico. METÁSTASIS A DISTANCIA MX: Metástasis a distancia no puede ser evaluada. M0: Sin evidencia de metástasis a distancia. M1: Presencia de metástasis a distancia. Estadiaje del cáncer de mama: Estadio 0 Estadio I Estadio IIA Estadio IIB Estadio
IIIA Estadio IIIB
Estadio IIIC
Estadio IV
TisN0M0 T1N0M0 T0N1M0 T2N1M0 T0N2M0 T4N0M0 Cualquier TN3M0
Cualquier T cualquier N M1
T1N1M0 T3N0M0 T1N2M0 T4N1M0 T2N0M0 T2N2M0 T4N2M0 T3N1M0 T3N2M0

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
PACIENTE E INVESTIGADOR: ?? FIRMAR Y FECHAR DOS EJEMPLARES DEL CONSENTIMIENTO INFORMADO. ?? GUARDAR UNO EN LA HISTORIA CLÍNICA. ?? ENTREGAR EL OTRO A LA PACIENTE. Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 66 de 82
APÉNDICE 4: MODELO DE CONSENTIMIENTO INFORMADO DE LA PACIENTE CONSENTIMIENTO INFORMADO DE PRESELECCIÓN. PRUEBA DEL TRIPLE NEGATIVO PARA EL ESTUDIO CIBOMA 2004-01 INTRODUCCIÓN Y OBJETIVOS Su médico está considerándola para participar en un estudio de investigación clínica (también conocido como “ensayo clínico”). Podrá participar en este ensayo una vez que haya completado todos los demás tratamientos que se le hayan administrado para el cáncer de mama que padece. Antes de que sigamos hablando de su posible participación en el estudio, se le deberán realizar otras pruebas en una muestra del tumor, conocidas como pruebas de los receptores hormonales y del HER2. Estas pruebas pueden llevarse a cabo con una muestra ya existente, de modo que no serán necesarias más biopsias, operaciones ni extracciones de sangre. Las pruebas de los receptores hormonales y del HER2 se realizan de forma rutinaria en el laboratorio de su hospital. Si el resultado es negativo, las pruebas se repetirán en un laboratorio central en Madrid, España, para confirmar el resultado negativo. El presente documento pretende proporcionarle la información suficiente para que usted comprenda los posibles riesgos y beneficios que pueden conllevar estas pruebas adicionales y su confirmación. Si después usted decide participar en el ensayo clínico, su médico le pedirá que lea y firme un documento que contiene más detalles acerca del ensayo. LA PRUEBA DE LOS RECEPTORES HORMONALES El tejido de la mama contiene unos receptores de hormonas, llamadas estrógenos y progesterona, que regulan muchos procesos relacionados con el sexo femenino. Aproximadamente un 70% de los cánceres de mama, presentan la presencia de estos receptores. En aproximadamente un 20% de los tumores, estos receptores hormonales no están presentes, lo cual se denomina como receptores negativos. En el caso de su tumor, su médico ha determinado mediante una técnica que se denomina tinción de inmunohistoquímica, que tiene receptores hormonales negativos, lo cual es una de las 2 condiciones necesarias para que pueda participar en este ensayo clínico. LA PRUEBA DEL HER2 El HER2 es una proteína que se produce en altos niveles en un 20% de los tumores de cáncer de mama. En el otro 80% aproximadamente, no se detectan altos niveles de esta proteína. Si usted pertenece a este último grupo (HER2 negativo), podrá participar en el ensayo clínico. El resultado negativo de los receptores hormonales y del HER2 deberá confirmarse en el laboratorio central. El material de la prueba se conservará en el laboratorio central para referencia en el futuro. Los laboratorios procesarán las muestras utilizando uno de los métodos siguientes:
- Tinción inmunohistoquímica (IHQ). - Hibridación por fluorescencia in situ (FISH).
PRUEBAS ADICIONALES Si entra a participar en el estudio CIBOMA 2004-01 en el futuro, se realizarán otras dos pruebas sobre el material enviado al laboratorio central, para determinar si su tumor produce niveles elevados de una proteína llamada citokeratina 5, y de otra proteína llamada HER1, también conocida como receptor de EGF. El proceso que se lleva a cabo para estas pruebas es la tinción inmunihistoquímica. BENEFICIOS POTENCIALES Es posible que no obtenga ningún beneficio personal a raíz de las pruebas adicionales realizadas en la muestra de tumor, aunque la información sobre los factores analizados podrá resultar útil para dirigir el tratamiento en el futuro. Su médico le facilitará el resultado de estas pruebas. COSTES ADICIONALES O COMPENSACIONES

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
PACIENTE E INVESTIGADOR: ?? FIRMAR Y FECHAR DOS EJEMPLARES DEL CONSENTIMIENTO INFORMADO. ?? GUARDAR UNO EN LA HISTORIA CLÍNICA. ?? ENTREGAR EL OTRO A LA PACIENTE. Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 67 de 82
Usted no tendrá que pagar la prueba de los receptores hormonales y del HER2, ni ningún otro test adicional. Tampoco se le pagará por acceder a que se realicen los análisis. CONFIDENCIALIDAD DE LOS DATOS Usted tiene derecho a la privacidad, y toda la información que se recoja a través de este estudio será confidencial en la medida que la ley lo permite. En España, el tratamiento de los datos de carácter personal se rige por la Ley Orgánica 15/1999, de 13 de diciembre. El consentimiento para el tratamiento de sus datos personales y para la cesión de los mismos es revocable. Usted puede ejercer el derecho de acceso, rectificación y cancelación dirigiéndose a su médico, el cual se pondrá en contacto con el promotor. Si los niveles de receptores hormonales y HER2 son positivos, y usted no puede participar en el ensayo clínico, su médico archivará en la historia clínica su consentimiento informado de preselección firmado y fechado, los resultados de las pruebas, y las observaciones médicas relacionadas con la preselección. Si los niveles de receptores hormonales y HER2 son negativos, usted podrá participar en el ensayo clínico CIBOMA 2004-01. En este caso, sus resultados serán recogidos por su médico y entregados a CIBOMA, Madrid, España, donde se encuentra la base de datos de investigación del estudio. Sus registros médicos originales podrán ser analizados durante o después del estudio por CIBOMA o sus representantes designados, las autoridades sanitarias y los representantes del Comité Ético local. Usted no tiene la opción de solicitar que no se analicen sus registros médicos. Si usted puede y decide participar en el estudio de CIBOMA, sus datos podrán ser analizados en cualquiera de los países miembros de la Coalición, para determinar cuántas pacientes presentan tumores con sus mismas características (los llamados triple negativo). CIBOMA podrá enviar los resultados a las autoridades sanitarias de los países de CIBOMA que participan en este estudio, comunicarlos en reuniones médicas, o publicarlos en revistas especializadas, para que otros médicos los conozcan. Si abandona el estudio, seguirá utilizándose la información recopilada sobre usted hasta ese momento. Podrá permitírseles a los representantes de CIBOMA, de las autoridades sanitarias y del comité ético local, que inspeccionen su historia clínica para asegurarse de que la información recogida es correcta. En estos casos, usted podrá ser identificada personalmente. Es posible que CIBOMA necesite volver a analizar los datos de preselección, y realizar nuevas pruebas estadísticas con ellos. Los resultados de este estudio pueden utilizarse para una futura investigación médica. Su participación en el estudio CIBOMA 2004-01 está descrita en otra hoja de consentimiento. OBTENCIÓN DE INFORMACIÓN ADICIONAL Le animamos a que haga las preguntas que desee en cualquier momento del estudio. Si se le presentan problemas o tiene más preguntas acerca de la preselección o del estudio, o de sus derechos como paciente, llame al Dr................................................ al teléfono ........................ BASES DE LA PARTICIPACIÓN La participación en los procedimientos de preselección del estudio CIBOMA/2004-01 es voluntaria. Si no desea participar, no perderá los beneficios a los que tienen derecho de todos modos ni sufrirá penalización alguna.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
PACIENTE E INVESTIGADOR: ?? FIRMAR Y FECHAR DOS EJEMPLARES DEL CONSENTIMIENTO INFORMADO. ?? GUARDAR UNO EN LA HISTORIA CLÍNICA. ?? ENTREGAR EL OTRO A LA PACIENTE. Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 68 de 82
DECLARACIÓN Y FIRMA DE LA PACIENTE
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE
MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES
HORMONALES Y HER2neu NEGATIVOS
Número de estudio: CIBOMA/2004-01 Yo ................................................................. He leído la hoja de información que se me ha entregado y entiendo el propósito de la selección para el análisis de la expresión de receptores hormonales y HER2. He podido hacer preguntas sobre el estudio He recibido respuestas satisfactorias a mis preguntas He recibido suficiente información sobre el estudio. He hablado con ...................................................... Comprendo que mi participación es voluntaria. Comprendo que puedo renunciar a mi participación en este proceso de selección: 1. Cuando quiera 2. Sin tener que dar explicaciones 3. Sin que esto repercuta en mis cuidados médicos Presto libremente mi conformidad para participar en esta selección para el análisis de receptores hormonales y HER2 en una muestra de mi cáncer de mama primario. Fecha Firma del participante: Yo he explicado por completo los detalles relevantes de este estudio al paciente nombrado anteriormente. Fecha: Firma del investigador:

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
PACIENTE E INVESTIGADOR: ?? FIRMAR Y FECHAR DOS EJEMPLARES DEL CONSENTIMIENTO INFORMADO. ?? GUARDAR UNO EN LA HISTORIA CLÍNICA. ?? ENTREGAR EL OTRO A LA PACIENTE. Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 69 de 82
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE
MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE,
RECEPTORES HORMONALES Y HER2neu NEGATIVOS
Número de estudio: CIBOMA/2004-01 Su médico le ha explicado que padece cáncer de mama con riesgo de recaída. Existen diversos tratamientos que pueden ayudarle. Le invitamos a que participe en un estudio de investigación del fármaco Xeloda? (capecitabina, en abreviatura llamado X), un fármaco oral que se ha mostrado muy eficaz en el tratamiento del cáncer de mama avanzado. El ensayo se está llevando a cabo en varios hospitales de América y España, y se espera la participación de 1324 pacientes en el marco de un estudio de investigación aprobado por las autoridades de su Hospital y de las autoridades sanitarias de su país. Este estudio está promovido por la Coalición Iberoamericana de Investigación en Oncología Mamaria (CIBOMA), asociación sin ánimo de lucro cuyo objetivo es aumentar la calidad y número de los ensayos clínicos en cáncer de mama. OBJETIVO El objetivo de este estudio es comprobar si la prolongación del tratamiento intravenoso de quimioterapia estándar, que ya ha recibido, con un tratamiento de quimioterapia oral (capecitabina), se muestra eficaz para prevenir la recaída de su enfermedad. Tendrá la misma posibilidad de recibir tratamiento con 8 ciclos de Xeloda? , que de no recibir tratamiento y ser asignada a un brazo de observación. Esto se decidirá al azar, como tirar una moneda al aire. DESARROLLO DEL ESTUDIO Antes de recibir el tratamiento, se le realizarán una serie de pruebas para determinar si puede participar en el estudio. Estas pruebas incluirán análisis de sangre y una exploración física. Es posible que deba realizarse una evaluación de su función cardiaca y procedimientos radiológicos para comprobar si su enfermedad no se ha extendido a otras partes del cuerpo. También realizaremos las determinaciones rutinarias en su tumor, tales como estado de los receptores hormonales y de una proteína denominada HER2. Para que estas determinaciones sean homogéneas, usted habrá aceptado previamente el envío de una muestra de su tumor a un laboratorio central, diferente del de su propio hospital. También es posible que su médico le solicite una muestra de sangre más, para analizar la relación entre sus características individuales y una mayor sensibilidad a los tratamientos de este estudio. Mientras permanezca en el estudio, y si le ha correspondido el brazo de tratamiento con capecitabina, se le realizarán análisis de sangre antes del inicio de cada nuevo ciclo (cada 21 días). Estos análisis de sangre regulares y otros exámenes se realizarán para comprobar que los fármacos no afectan de manera adversa a la médula ósea, riñones e hígado. Si se le asigna al brazo A de tratamiento (capecitabina) deberá recibir el tratamiento durante 24 semanas (es decir, 8 ciclos de 21 días). Cada ciclo de tratamiento consiste en dos semanas de tratamiento seguidas de una semana de descanso. Si se le asigna al brazo B (observación), no recibirá tratamiento, pero su médico le pedirá que acuda a visitarse de forma rutinaria. Posteriormente, su médico le realizará un seguimiento al final del estudio. A continuación, el médico realizará su seguimiento del mismo modo que en las demás pacientes con cáncer de mama, con el fin de confirmar que el tumor no ha presentado recurrencia.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
PACIENTE E INVESTIGADOR: ?? FIRMAR Y FECHAR DOS EJEMPLARES DEL CONSENTIMIENTO INFORMADO. ?? GUARDAR UNO EN LA HISTORIA CLÍNICA. ?? ENTREGAR EL OTRO A LA PACIENTE. Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 70 de 82
Durante los 2 primeros años, se le realizará una exploración física cada tres meses, y se realizará una mamografía y una radiografía de tórax cada 12 meses. Durante los años 3 a 5, se suprimirán las visitas cada 3 meses, pero se seguirán realizando las visitas cada 6 y 12 meses con las mismas evaluaciones. Se le pedirá que rellene un cuestionario sobre calidad de vida antes del inicio de la quimioterapia, antes de cada ciclo de tratamiento y durante el período de seguimiento (6, 12 y 24 meses después del final de la quimioterapia), para saber qué piensa del tratamiento administrado para su enfermedad. Este procedimiento ayudará a sus médicos a determinar la eficacia, evaluada por medios clínicos, frente al beneficio que usted experimenta. El fármaco Xeloda? se administrará por vía oral, dos veces al día (mañana y noche) con agua dentro de los 30 minutos posteriores a una comida. Deberá seguir el tratamiento durante 14 días ininterrumpidos, tras lo cual lo interrumpirá durante una semana, para volver a reanudarlo posteriormente hasta completar el tratamiento. POSIBLES ACONTECIMIENTOS ADVERSOS Los siguientes son los efectos secundarios más frecuentes del fármaco empleado en este estudio. Existen efectos secundarios que conocemos en este momento. Sin embargo, puesto que este es un estudio sobre nuevos tratamientos podrían existir otros efectos secundarios que desconocemos todavía. Por tanto, es muy importante que usted informe inmediatamente a su médico acerca de cualquier síntoma inusual. Con Xeloda? , usted podría experimentar diarrea, dolor abdominal, náuseas, irritación bucal que la molestaría al comer y el denominado síndrome mano-pie, que consiste en un entumecimiento de manos y pies que puede ir acompañado de dolor, descamación de la piel y malestar que puede afectar a sus tareas diarias. También podrían alterarse los niveles de calcio en su sangre, pero su médico controlará regularmente los mismos. Estos efectos secundarios podrían ser pequeños inconvenientes o podrían ser severos, pero su médico la vigilará de cerca y si ocurriera algo decidirá ajustar su dosis de quimioterapia o parar el tratamiento. En el caso de que se suspendiera el tratamiento, se le ofrecería a usted un tratamiento alternativo. En caso de que experimente fiebre o hematomas después de recibir cualquier fármaco, deberá contactar con los médicos del departamento. BENEFICIOS ESPERADOS Usted ya ha recibido un tratamiento con quimioterapia intravenosa estándar previo a este estudio. Es posible que también haya recibido tratamiento con radioterapia. Con cualquiera de los tratamientos estándar, hay un porcentaje importante de mujeres que se curan completamente de su cáncer de mama. Este estudio nos ayudará a saber si prolongar el tratamiento con quimioterapia aumenta este porcentaje de pacientes que se curan completamente, o por el contrario, esto no sucede. Es decir, el cáncer de mama recurre, en forma de metástasis, en el mismo porcentaje de mujeres que han recibido el tratamiento estándar. Si este fuera su caso, su médico se encargará de proponerle el mejor tratamiento disponible para usted en ese momento. Su participación en este estudio puede, por tanto, no reportarle ningún beneficio a usted, pero nos ayudará a conocer mejor la enfermedad, y esperamos que esta información nos sirva para ayudar a mujeres que desarrollen la enfermedad en los próximos años. TRATAMIENTOS ALTERNATIVOS DISPONIBLES En estos momentos no se considera ningún tratamiento alternativo disponible en su situación y para su tipo de tumor. Lo habitual es que no reciba más tratamientos, y que se le cite para acudir a

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
PACIENTE E INVESTIGADOR: ?? FIRMAR Y FECHAR DOS EJEMPLARES DEL CONSENTIMIENTO INFORMADO. ?? GUARDAR UNO EN LA HISTORIA CLÍNICA. ?? ENTREGAR EL OTRO A LA PACIENTE. Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 71 de 82
sus revisiones rutinarias, que es lo que ocurrirá si se le asigna al brazo de observación de este estudio. SEGURO DEL ESTUDIO Para el caso de que se le ocasione algún perjuicio durante el estudio, CIBOMA ha suscrito una póliza de seguros con la compañía Gerling-Konzern, que cubre la responsabilidad civil del promotor del estudio, de los investigadores principales y sus colaboradores así como del titular del Hospital o Centro en los términos que se establecen en el Real Decreto 223/2004, que regula la realización de ensayos clínicos con medicamentos en España. Se le informará de los hallazgos nuevos significativos de Xeloda? que se descubran durante el estudio y que pueden hacer que cambie su intención de participar en el mismo. No debe tomar parte en este estudio si piensa que está embarazada o que existe la posibilidad de que pueda quedarse embarazada durante el estudio. Por consiguiente, antes de iniciar el tratamiento, su médico comprobará que está utilizando un método anticonceptivo eficaz. Su médico puede decidir su exclusión del estudio si es perjudicial para usted, si no sigue las instrucciones del tratamiento, se descubre que no cumple los requisitos del ensayo o si se suspende el estudio. ASPECTOS ECONÓMICOS Debe usted saber que su médico cobrará unos honorarios por su participación en este ensayo clínico. Sin embargo, no está previsto que se le abonen a usted honorarios por su propia participación, ni por el gasto extra que pueda suponerle la misma por desplazamientos al hospital u otros conceptos. CARÁCTER VOLUNTARIO DE SU PARTICIPACIÓN Su participación en este estudio es voluntaria. Si decide participar, pero posteriormente cambia de opinión, es libre de hacerlo y no tiene que aducir ningún motivo. Sin embargo, debe informar a su médico de su decisión, de manera que pueda indicarle el procedimiento que se debe seguir para evaluar adecuadamente su patología clínica y, a continuación, proseguir con los cuidados médicos. La asistencia médica que reciba de su médico no se verá afectada por su decisión. PERSONAS CON ACCESO A SUS DATOS Y PROTECCIÓN DE LA CONFIDENCIALIDAD Si participa en el estudio, su historia clínica podrá ser revisada por el personal profesional de CIBOMA, las Autoridades Sanitarias o los Organismos internacionales pertinentes. Los datos derivados de su participación en el estudio se pueden publicar con fines científicos, pero su identidad se mantendrá de forma confidencial, es decir, nadie podrá conocer su identidad a partir de los datos publicados. Se garantizará, en todo caso, el respeto a la confidencialidad de sus datos personales, de acuerdo con la normativa vigente en su país. En el caso de que se plantee algún problema o pregunta con relación a este estudio, sus derechos como participante en una investigación clínica o cualquier perjuicio relacionado con la investigación, debe contactar con: Dr:....................................................................tel:.............................................................

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
PACIENTE E INVESTIGADOR: ?? FIRMAR Y FECHAR DOS EJEMPLARES DEL CONSENTIMIENTO INFORMADO. ?? GUARDAR UNO EN LA HISTORIA CLÍNICA. ?? ENTREGAR EL OTRO A LA PACIENTE. Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 72 de 82
CONSENTIMIENTO DEL PACIENTE POR ESCRITO
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE
MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES
HORMONALES Y HER2neu NEGATIVOS
Número de estudio: CIBOMA/2004-01 Yo ................................................................. He leído la hoja de información que se me ha entregado He podido hacer preguntas sobre el estudio He recibido respuestas satisfactorias a mis preguntas He recibido suficiente información sobre el estudio. He hablado con ...................................................... Comprendo que mi participación es voluntaria. Comprendo que puedo retirarme del estudio: 1. Cuando quiera 2. Sin tener que dar explicaciones 3. Sin que esto repercuta en mis cuidados médicos Presto libremente mi conformidad para participar en el estudio. Fecha Firma del participante: Fecha: Firma del investigador:
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 73 de 82
APÉNDICE 5: HOJA DE NOTIFICACION DE LOS ACONTECIMIENTOS ADVERSOS
NOTIFICACION DE SOSPECHA DE REACCION ADVERSA PARA
MEDICAMENTOS EN INVESTIGACIÓN
CODIGO DE PROTOCOLO (promotor)
Nº NOTIFICACION (Promotor)
PACIENTE Nº Nº NOTIFICACION
I. INFORMACION SOBRE LA REACCIÓN ADVERSA 2. FECHA DE NACIMIENTO 2a.
EDAD 3. SEXO 3a.
PESO 3b. TALLA
4-6. FECHA DE INICIO DE LA REACCIÓN
1a. PAÍS
DÍA
MES
AÑO
? HOMBRE ? MUJER
DÍA
MES
AÑO
7. DESCRIPCIÓN DE LA REACCIÓN ADVERSA (Incluyendo resultados relevantes de exploración o de laboratorio, y la fecha de finalización, si procede).
8-13b. CRITERIOS DE GRAVEDAD/ DESENLACE ? FALLECIMIENTO ? LA VIDA DEL PACIENTE HA ESTADO EN PELIGRO ? HOSPITALIZACIÓN ? PROLONGACIÓN HOSPITALIZACIÓN ? INCAPACIDAD PERMANENTE O SIGNIFICATIVA ? RA CLINICAMENTE RELEVANTE ? PERSISTENCIA DE LA REACCIÓN ADVERSA ? RECUPERACIÓN
II. INFORMACION DEL MEDICAMENTO EN INVESTIGACIÓN 14. MEDICAMENTO SOSPECHOSO
15. DOSIS DIARIA
16. VÍA 17. ENFERMEDAD EN ESTUDIO
18. FECHAS DE INICIO FINAL
19. DURACIÓN DEL TRATAMIENTO
20. ¿REMITIÓ LA REACCIÓN AL SUSPENDER LA MEDICACIÓN? ? SI ? NO ? NO PROCEDE
20a. ¿REMITIÓ LA REACCIÓN AL REDUCIR LA DOSIS? ? SI ? NO ? NO PROCEDE
21. ¿REAPARECIÓ LA REACCIÓN AL ADMINISTRAR DE NUEVO LA MEDICACIÓN? ? SI ? NO ? NO PROCEDE

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 74 de 82
III. MEDICAMENTOS CONCOMITANTES E HISTORIA CLÍNICA 22. MEDICAMENTOS CONCOMITANTES (Márquese con un asterisco el o los medicamentos sospechosos)
22a. DOSIS DIARIA
22b. VÍA
22c. FECHAS DE INICIO FINAL
22d. MOTIVO DE LA PRESCRIPCIÓN
23. DATOS IMPORTANTES DE LA HISTORIA CLÍNICA (ej. diagnósticos, alergias, embarazos, etc.) IV. INFORMACION SOBRE PROMOTOR E INVESTIGADOR
24a. NOMBRE Y DIRECCION DEL PROMOTOR
24b. NOMBRE Y DIRECCION DEL INVESTIGADOR
24c. CODIGO DE LABORATORIO (Nº AEM)
25a. TIPO DE INFORME
? INICIAL ? SEGUIMIENTO
24c. TECNICO DEL PROMOTOR QUE INFORMA NOMBRE: TELEFONO: FIRMA:
24e. FECHA DEL INFORME
24f. FECHA DE ENTRADA AEM
25b. SE ADJUNTA INFORME COMPLEMENTARIO

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 75 de 82
APÉNDICE 6: LISTA DE PAÍSES PARTICIPANTES En España: Los siguientes investigadores han manifestado su interés por participar en el presente estudio. Su incorporación al mismo se realizará de forma escalonada. También está previsto que participen en el estudio centros de Argentina, Brasil, Chile, Guatemala, Méjico, Perú, Uruguay y Venezuela. INVESTIGADOR HOSPITAL
Jesús García Mata H. Sta. María Nai
José Ramón Mel Lorenzo H. Xeral Calde
Laura de Paz H. Arquitecto Marcide
Javier Castellanos H. Xeral Cíes
Manuel Constela H. Montecelo
Manuel Ramos Centro Oncológico Galicia
Lourdes Calvo H. Juan Canalejo
Concepción Almanza Madera Policlínico de Vigo POVISA
Dolores Menendez Prieto C.Hospit.Univ. Santiago
Cristina Martín H. Del Espiritu Santo
Agustín Barnadas i Molins H. de la Santa Creu i Sant Pau
Sonia González H. Mutua Terrassa
Rosa Mª Franquesa Grané H. General de VIC
Miguel Angel Segui Hospital Parc Taulí
Montserrat Muñoz Mateu H. Clínico y Provincial
Alfonso Modolell Ins. Oncológico Corachán
Amadeu Pelegrí Hospital Sant Joan de Reus
Miguel Gil Instituto Catalán de Oncología
Angels Arcusa Lanza Consorci Sanitari de Terrassa
Ignacio Tusquets Trias de Bes Hospital del Mar
Mireia Margelí H. Germans Trias i Puyol
Ramón Colomer Bosch H. Josep Trueta
Isabel Moreno Solorzano H. Municipal de Badalona
Ana de Miguel H. San Juan de Dios de Manresa
Ana Balill i Gilart H. Arnau de Vilanova
Montserrat LLobera H. Tortosa Verge de la Cinta
Montserrat Boleda H. Sant Camil
Antonio Fernández Aranburo Hospital General de Albacete
Javier Cassinello H. Gral. de Guadalajara
Miguel Ángel de la Cruz Mora H. Virgen de la Salud
Pedro Puñal H. Provincial de Toledo
Mª del Mar Muñoz H. Virgen de la Luz
Encarna Adrover H.Gral. Univ. de Alicante
Mª José Godes Sanz Bremond H. General de Valencia
Vicente Carañana H. Arnau de Vilanova
Amparo Ruiz Simón I.V.O.
Alvaro Rodriguez Lescure H. General de Elche
José Miguel Cuevas Hospital de la Ribera
Ana Lluch H. Clínico de Valencia
Antonio Galán Brotons H. Puerto de Sagunto
Miguel Pastor Borgoñón H. Universitario la Fe
Santiago Olmos H. Universitario Dr. Peset

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 76 de 82
Cristina Llorcas H. General de Elda
Adolfo Frau Llopis H. Provincial Castellón
José Lizón Giner H. Clínico San Juan
Mª Dolores Torregrosa H. Lluis Alcanys
Amparo Oltra H. Virgen de los Lirios
Norberto Batista H. Universitario de Canarias
Adolfo Murias Rosales Hospital Insular
José Aguiar Morales H. Gran Canaria Dr. Negrin
Javier Dorta C. Hosp. Ntra. Sra. De la Candelaria
José Ignacio Mayordomo H. Clinico Univ. de Zaragoza
Jesús Florián Jericó H. Comarcal Barbastro
Antonio Antón Torres H. Miguel Servet
Alfonso Yubero H. Gral de Teruel O. Polanco
Miguel Martin Jimenez H. Clínico San Carlos
Ricardo Cubedo Clínica Puerta de Hierro
Miguel Ángel Lara Álvarez H. Severo Ochoa
Miguel Méndez Ureña H. General de Móstoles
José Enrique Alés Martínez H. Ruber Internacional
Carlos Jara Sánchez Fundación Hosp. Alcorcón
Eduardo García Rico Hospital de Madrid
Eduardo García Rico Hospital de Montepríncipe
Gumersindo Pérez Manga H. Gregorio Marañón
César Mendiola Fernández H. Doce de Octubre
Carmen Crespo H. Ramón y Cajal
Francisco Lobo Samper Fundación Jimenez Díaz
Santos Enrech Francés Hospital Gomez Ulla
Amalia Velasco Ortiz de Taranco Hospital la Princesa
Ramón Pérez Carrión Clínica MD. Anderson
Javier Salvador Bofill H. U. De Valme
Manuel Ruiz Borrego H. Univ. Virgen de Rocio
José Mª Baena Cañada H. Puerta del Mar
Enrique Aranda Aguilar H. Reina Sofía
Pedro Sánchez Rovira H. Gral. Especalid. Jaén
Liliana Canosa Hospital Torrecárdenas
Antonio Lorenzo Peñuelas H. Univ. Puerto Real
Joaquín Belón H. Virgen de la Nieves
Encarnación Jimenez H.Gral de Jerez Frontera
Juan Lucas Bayo Calero H. Juan Ramón Jimenez
Pedro Valero Jimenez Clínica Sagrado Corazón
Rafael Trujillo Hospital Punta de Europa
José Luis García Puche H. Clínico San Cecilio
Nuria Ribelles H. U. Virgen de la Victoria
Francisco Carabantes C.H. Carlos Haya
Arrate Plazaola Alcibar Inst. Oncológico Guipuzcoa
Isabel Alvarez López Hospital Donostia
J. Ramón Barceló Hospital de Cruces
Purificación Martinez Prado Hospital de Basurto
Severina Dominguez Hospital Txagorritxu
José Luis Alonso Romero H. Virgen de la Arrixaca
Francisco Ayala de la Peña H. Morales Meseguer

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 77 de 82
José Manuel López Vega Hops. U. Marques Valdecilla
López Lara H. Clínico Univ. Valladolid
Alberto Arizcum Hospital Rio Carrión
César Rodriguez Sánchez H. Universitario Salamanca
García Palomo H. General de León
Yolanda Fernández H. General Yague
José Valero Alvarez Gallego H. Rodriguez Chamorro
Juan Carlos Torrego H. Univ. del Rio Hortega
Florestán Juarez H. Ntra. Senora del Prado
Mª Lomas Garrido Hospital Infanta Cristina
Pablo Borrega H. San Pedro Alcantara
Yolanda López del Puerto H. Virgen del Puerto
Mª Ángeles Panadero H. Ciudad de Coria
Antón Avellá H.Son Dureta
José Juan Irramendi Mañas Hospital de Navarra
Edelmira Velez de Mendizabal C. Hosp. San Millan-San Pedro

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 78 de 82
APÉNDICE 7: REGISTRO DE ESTADO MENSTRUAL TABLA DE REGISTRO DE ESTADO MENSTRUAL
INSTRUCCIONES: Utilice S si hay sangrado pero no se necesita protección sanitaria. Utilice C si se necesita protección sanitaria (compresas, tampones, proteje -slips, etc). Déjelo en blanco si no hay sangrado.
Fecha de Comienzo del Registro (M, D, A) 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 Ene Feb Mar Abr May Jun Jul Ago Sep Oct Nov Dic COMENTARIOS:____________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________ _____________________________________________
Por favor, ponga su nombre en letras de imprenta INVESTIGADOR: Guarde una copia para sus archivos.

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 79 de 82
APÉNDICE 8: CRITERIOS DE COCKROFT Y GAULT Fórmula de Cockroft-Gault para mujeres: Aclaramiento de creatinina (ml/min) = [(140 – edad) x peso corporal actual (en Kg) x 0.85] / [72 x creatinina sérica (en mg/dL)] o [(140 – edad) x peso corporal actual (en Kg.) x 0.85] / [0.81 x creatinina sérica (en ?mol/L)]
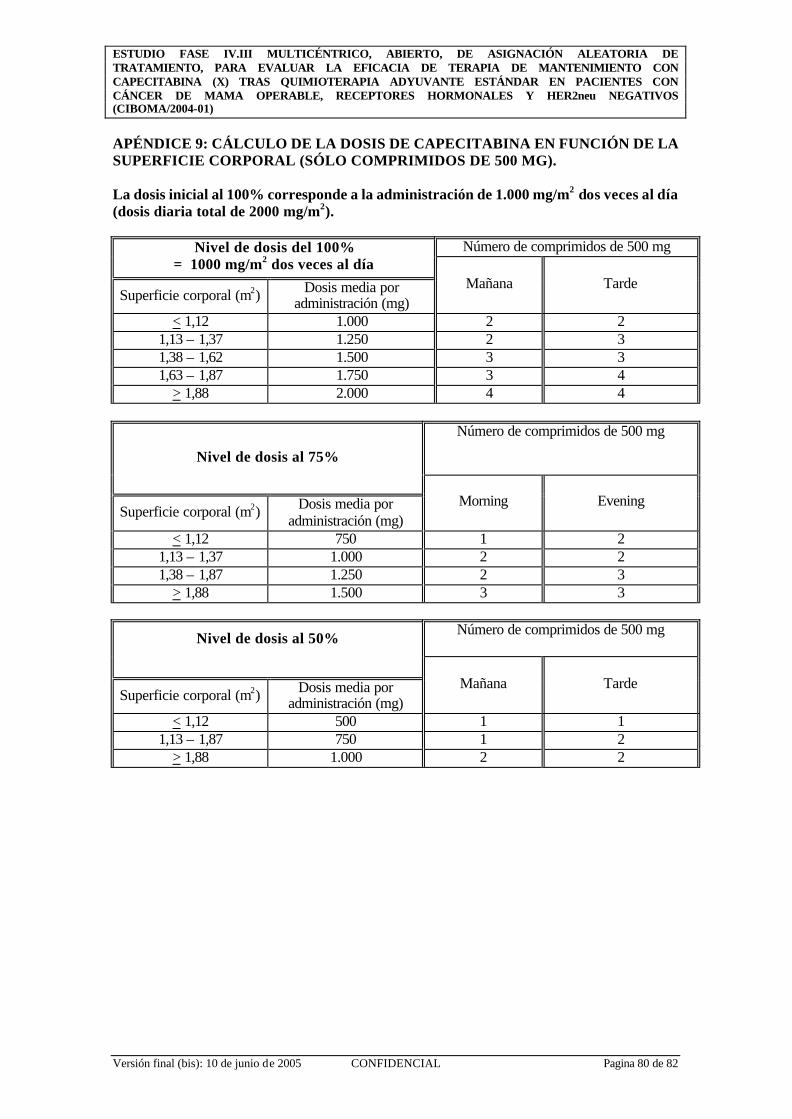
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 80 de 82
APÉNDICE 9: CÁLCULO DE LA DOSIS DE CAPECITABINA EN FUNCIÓN DE LA SUPERFICIE CORPORAL (SÓLO COMPRIMIDOS DE 500 MG). La dosis inicial al 100% corresponde a la administración de 1.000 mg/m2 dos veces al día (dosis diaria total de 2000 mg/m2).
Número de comprimidos de 500 mg Nivel de dosis del 100% = 1000 mg/m2 dos veces al día
Superficie corporal (m2) Dosis media por administración (mg)
Mañana Tarde
< 1,12 1.000 2 2 1,13 – 1,37 1.250 2 3 1,38 – 1,62 1.500 3 3 1,63 – 1,87 1.750 3 4
> 1,88 2.000 4 4
Número de comprimidos de 500 mg
Nivel de dosis al 75%
Superficie corporal (m2) Dosis media por administración (mg)
Morning Evening
< 1,12 750 1 2 1,13 – 1,37 1.000 2 2 1,38 – 1,87 1.250 2 3
> 1,88 1.500 3 3
Número de comprimidos de 500 mg Nivel de dosis al 50%
Superficie corporal (m2) Dosis media por administración (mg)
Mañana Tarde
< 1,12 500 1 1 1,13 – 1,87 750 1 2
> 1,88 1.000 2 2
ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 81 de 82

ESTUDIO FASE IV.III MULTICÉNTRICO, ABIERTO, DE ASIGNACIÓN ALEATORIA DE TRATAMIENTO, PARA EVALUAR LA EFICACIA DE TERAPIA DE MANTENIMIENTO CON CAPECITABINA (X) TRAS QUIMIOTERAPIA ADYUVANTE ESTÁNDAR EN PACIENTES CON CÁNCER DE MAMA OPERABLE, RECEPTORES HORMONALES Y HER2neu NEGATIVOS (CIBOMA/2004-01)
Versión final (bis): 10 de junio de 2005 CONFIDENCIAL Pagina 82 de 82
APÉNDICE 10: INTERRUPCIÓN Y EXTENSIÓN DEL PERIODO DE REPOSO EN EL TRATAMIENTO CON CAPECITABINA
Tratamiento normal : Día 1
Interrupciones durante el t ratamiento:
Las interrupciones se consideran dosis perdidas. Se mantendrá el p lan de tratamiento in ic ia l .
Extensión de los per iodos de descanso:
I se ext iende un per iodo de descanso debido a toxic idad, se administrará el “c ic lo" completo poster iormente.
7 días 14 días 14 días
Día 8
Día 15
Día 22
Día 29
Día 36
Día 4 3
Día 50