guia para memoria e informes master bioinfo v2 · un inhibidor en una enzima), o moléculas de agua...
TRANSCRIPT

CASP 11 EDUARDO ROMEU GRACIA
MÁSTER EN BIOINFORMÁTICA Y BIOLOGÍA COMPUTACIONAL ESCUELA NACIONAL DE SALUD- INSTITUTO DE SALUD CARLOS III
2013-2014
CENTRO NACIONAL DE INVESTIGACIONES ONCOLOGICAS
DIRECTOR DE LA TESIS: MICHAEL TRESS
CODIRECTOR DE LA TESIS: FLORENCIO PAZOS
FEBRERO DE 2015

Índice:
1 Objetivo Pág. 1
2 Introducción " 1
2.1 CASP " 1
2.1.1 Formato PDB " 3
2.1.2 Puntuación en CASP " 4
2.2 Utilidad " 6
2.3 Estructuras limitadas " 8
2.4 Diversas estructuras " 8
2.4.1 Dominios " 8
2.4.2 Péptido señal " 12
2.4.3 Expression tag " 13
2.4.4 Regiones desordenadas " 13
2.4.5 Regiones transmembrana " 14
2.4.6 Puentes disulfuro " 15
2.4.7 Coiled coils " 16
2.4.8 Otras estructuras " 16
2.5 Vocabulario " 17
3 Material y métodos " 18
3.1 Bases de datos " 18
3.1.1 PDB " 18
3.1.2 SCOP y CATH " 18
3.1.3 PFAM " 19
3.1.4 UniProt " 20
3.2 Clasificación de métodos de predicción " 20
3.3 Métodos de predicción " 20
3.4 Búsqueda de secuencias " 21
3.4.1 BLAST " 22
3.4.2 PsiBLAST " 23
3.4.3 Jackhmmer " 23
3.4.4 HHBlits " 24
3.4.5 Número de secuencias " 24
3.5 Búsqueda de templates " 24
3.5.1 Si no hay template o es remoto " 27
3.6 Alineamientos " 27
3.6.1 Jalview " 28
3.7 Predicción de contactos " 29
3.7.1 L-factor " 30
3.8 Métodos de predicción de estructura secundaria " 30
3.8.1 PSIPRED " 30
3.8.2 JPRED " 31
3.8.3 GeneSilico " 31
3.9 Programas para generar modelos " 32
3.9.1 Modeller " 32
3.9.2 FoldIt " 36
3.9.3 Swiss Model " 37
3.9.4 Ev Fold " 37
3.10 Visualización de modelos " 37
3.11 Elección del mejor modelo " 39
4 Resultados " 41
5 Discusión " 47
6 Conclusión " 48
7 Bibliografía " 49

1. OBJETIVO
En mis prácticas, las cuales han tenido una duración de 3 meses, ocupando los meses de mayo,junio y julio, he participado en una competición internacional conocida como CASP cuya finalidades la predicción de la estructura de diversas proteínas. Este año se ha llevado a cabo la decimoprimera edición de este “concurso” en el que hemos intervenido Michael Tress, Juan Rodriguez yun servidor como componentes del grupo de predicción que ha representado al CNIO.
El objetivo es muy sencillo, debemos hacer uso de todos los programas, bases de datos, papers ydemás dispositivos e información de la mejor manera posible para generar unos buenos modelosestructurales de proteínas.
Para este propósito hemos trabajado en equipo repartiendo las diversas tareas llevadas a cabo yponiéndolas en común posteriormente para obtener un satisfactorio resultado para cada proteína.
2. INTRODUCCIÓN
2.1 CASP
CASP, cuyas siglas significan Critical Assessment of protein Structure Prediction, es unexperimento que se realiza cada dos años desde 1994 y cuyo principal objetivo es evaluar lastécnicas actuales en la predicción de estructura de proteínas. Lo que se pretende con CASP espromover los diferentes métodos existentes a la hora de identificar la estructura tridimensional deuna proteína.
Las proteínas que entran en concurso son proteínas con estructura resuelta por cristalografía derayos X o por espectroscopía de resonancia magnética nuclear (RMN) y cuyos datos no han sidointroducidos en la Protein Data Base (PDB) o base de estructuras de proteínas.
Se podría decir que en CASP participan dos grandes sectores, el grupo de predicción humana y elgrupo formado por servidores automáticos. Nuestro grupo (CNIO), como es evidente, haparticipado junto a uno de los grupos pertenecientes al sector de predicción humana.En total se han lanzado 100 targets, de los cuales hemos predicho la estructura de 55 (un total de 78dominios), que en principio son los más complicados de resolver y participan tanto humanos comoservidores, el resto de targets son únicamente para los servidores automáticos.
Todos los targets se iban colgando periódicamente en una página web: http://predictioncenter.org/casp11/index.cgi#targets
Como ya he mencionado, la finalidad es evaluar los métodos de predicción pero también se tomaeste experimento como una especie de competencia de los servidores entre sí y lo mismo pasa conlos grupos humanos. Al final de CASP se realizan unos rankings donde se muestran las posiciones de los diferentesgrupos y servidores que han participado según la puntuación obtenida en sus predicciones.
A la hora de trabajar con CASP, entrábamos en la página web mencionada y nos dirigíamos alapartado de “Targets”.
1
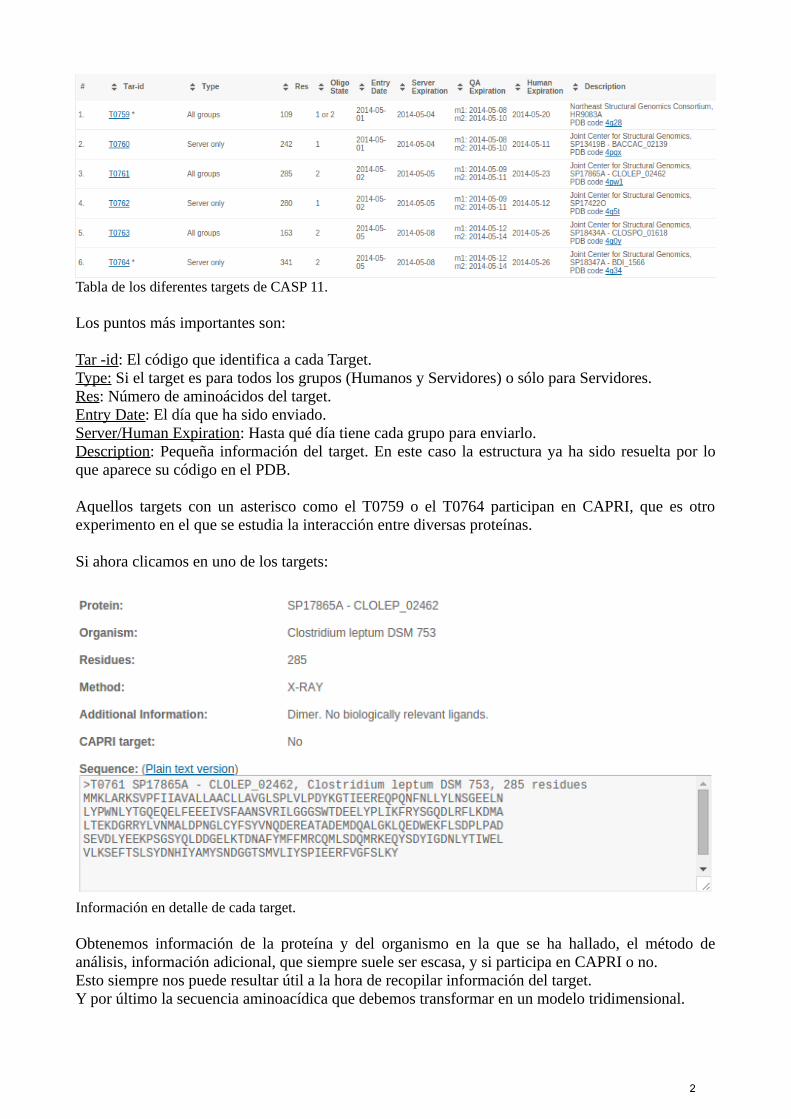
Tabla de los diferentes targets de CASP 11.
Los puntos más importantes son:
Tar -id: El código que identifica a cada Target.Type: Si el target es para todos los grupos (Humanos y Servidores) o sólo para Servidores.Res: Número de aminoácidos del target.Entry Date: El día que ha sido enviado.Server/Human Expiration: Hasta qué día tiene cada grupo para enviarlo.Description: Pequeña información del target. En este caso la estructura ya ha sido resuelta por loque aparece su código en el PDB.
Aquellos targets con un asterisco como el T0759 o el T0764 participan en CAPRI, que es otroexperimento en el que se estudia la interacción entre diversas proteínas.
Si ahora clicamos en uno de los targets:
Información en detalle de cada target.
Obtenemos información de la proteína y del organismo en la que se ha hallado, el método deanálisis, información adicional, que siempre suele ser escasa, y si participa en CAPRI o no.Esto siempre nos puede resultar útil a la hora de recopilar información del target.Y por último la secuencia aminoacídica que debemos transformar en un modelo tridimensional.
2

2.1.1 Formato PDB
Una vez tenemos la estructura completada debemos enviársela a los asesores del CASP en unformato muy semejante al formato PDB, el cuál tiene este aspecto:
Este es tan sólo un fragmento del código entero, pues en este caso era de 5700 líneas como éstas. Acontinuación detallo el significado de las columnas de datos más significativas::
La primera columna nos indica si estamos tratando con un átomo (ATOM) de la cadenapolipeptídica o si, por el contrario, se trata de un átomo que forma parte de un ligando ( por ejemploun inhibidor en una enzima), o moléculas de agua o átomos metálicos como hierro, cobre, zinc …apareciendo entonces la palabra “HETATM”.
La segunda columna indica el número del átomo.
La tercera columna describe a qué átomo nos referimos, basándose en la nomenclatura PDB: N es elnitrógeno del grupo amida, CA es el carbono alfa, C es el carbono carbonilo, O es el oxígenocarbonílico. CB es el carbono beta, CG carbono gama y CD1 y CD2 son los carbonos delta 1 y 2.
En la sexta columna se anota el número de residuo. Como vemos en el ejemplo, el último átomopertenece a un nuevo residuo.
Las columnas 7, 8 y 9 indican las coordenadas x, y, z.
La última columna indica el tipo de átomo, en términos generales (N, nitrógeno; H, Hidrógeno; O,Oxígeno ...)
De esta manera, con toda esta información, podemos representar mediante un código, una estructuratridimensional de una proteína.
3

2.1.2 Puntuación en CASP
Para el análisis de los resultados se emplea el método LGA (Local-Global Alignment) el cual estádiseñado para comparar estructuras de proteínas o fragmentos de las mismas y encontrar lassimilitudes tridimensionales [1]. El programa analiza las estructuras tridimensionales proteicas yrealiza la mejor superposición posible, alineando la mayor región que se puede superponer ycalculando posteriormente el GDT-TS.
El GDT-TS (Global Distance Test- Total Score) es el método que se usa para puntuar laspredicciones el cual mide la distancia de los carbonos alfa entre la proteína real y su predicción unavez superpuestas. A menor distancia, más semejante son ambas y por lo tanto la puntuación esmayor.
La ecuación empleada es la siguiente:GDT-TS = (GDT_P1 + GDT_P2 + GDT_P4 + GDT_P8)/4
Donde Pn representa el porcentaje de residuos a una distancia menor a n Å.
Pero aparte del GDT-TS que se basa únicamente en los carbonos alfa, existen otros métodos comoel GDT_HA, que tiene en cuenta todos los átomos y es muy útil para los TBM easy; el métodoCAD, que cuantifica las diferencias entre áreas de contacto; u otros muchos métodos más basadosen medidas estructurales.
La puntuación total obtenida con este cálculo oscila entre 0 y 100, siendo 100 un parecido absolutocon la estructura real.
El resultado final va a depender de si estamos tratando con un TBM o con un TFM:– Los Template Based Modeling (TBM) – A esta categoría pertenecen los modelos los cuales
tienen un template (o estructura proteica resuelta en el PDB) que abarca una parte o inclusola totalidad de nuestra proteína problema o target. Es decir, que en la base de datos del PDB,se encuentra una proteína con una estructura muy semejante y que puede servirnos de moldea la hora de generar nuestra estructura. En este caso se evaluará la alineación, el backbone yla colocación de las cadenas laterales.
– Los Template Free Modeling (FM) – Son aquellas proteínas que no disponen de un templateen el que basarse a la hora de resolver la estructura del target.
Si es TBM, al haber un template, la predicción es más sencilla y las puntuaciones son mayores, sinembargo, si nos enfrentamos a un TFM los resultados son bastante menores.En principio se dice que si la puntuación GDT-TS, en la mayoría de grupos, es superior a 50 esporque es un TBM con un template sencillo, si la puntuación está entre 30 y 50 es porque hay untemplate pero está algo más alejado y es más difícil de encontrar, y si la puntuación es menor de 30suele ser porque es TFM. Pero en realidad esto no es para nada exacto y son finalmente los asesoreslos que deciden si un target es TBM (easy o hard) o FM.
Por puro azar se puede conseguir un GDT-TS de 15, pues siempre va a acabar alineando aunque seauna mínima región con un alfa-hélice o un strand (lámina-beta).
Los datos resumidos de todos los grupos para un mismo target se representan mediante el siguientetipo de gráficas conocidas como GDT-plots:
4
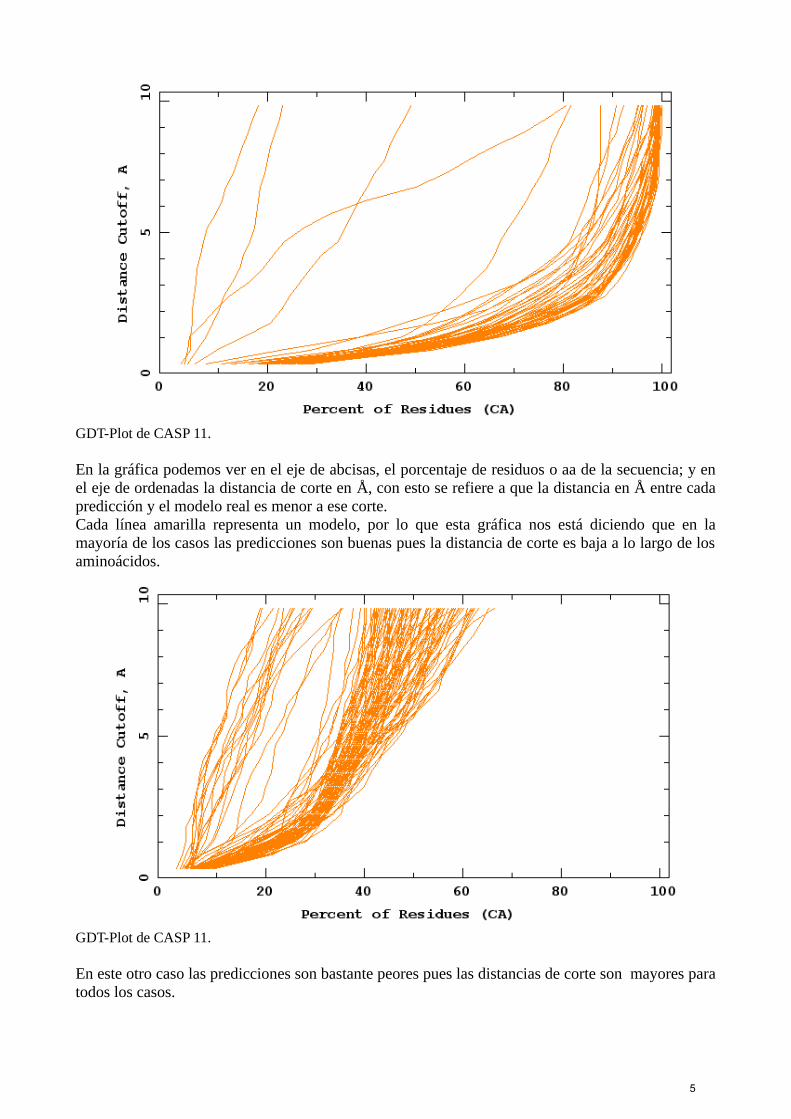
GDT-Plot de CASP 11.
En la gráfica podemos ver en el eje de abcisas, el porcentaje de residuos o aa de la secuencia; y enel eje de ordenadas la distancia de corte en Å, con esto se refiere a que la distancia en Å entre cadapredicción y el modelo real es menor a ese corte.Cada línea amarilla representa un modelo, por lo que esta gráfica nos está diciendo que en lamayoría de los casos las predicciones son buenas pues la distancia de corte es baja a lo largo de losaminoácidos.
GDT-Plot de CASP 11.
En este otro caso las predicciones son bastante peores pues las distancias de corte son mayores paratodos los casos.
5

2.2 Utilidad
Como ya he mencionado, mi labor se ha centrado en el uso de diversos métodos para predecirestructuras proteicas tridimensionales. Pero la pregunta que uno se hace después de esto es ¿paraqué nos puede servir conocer la estructura de las proteínas?, es decir, ¿qué finalidad tiene o quéempresas pueden estar interesadas?
Pues a modo de resumen podemos decir que puede ser muy útil en diversos campos de labiomedicina, ya que podemos asignar diversas proteínas a diferentes familias conociendo susparecidos y diferencias estructurales entre sí, o localizar diferentes puntos estructurales en unaproteína donde nos puede interesar que actúe un fármaco concreto [2].
Para concretar algo más, tenemos que destacar que según la precisión o la calidad del modeloobtenido puede ser útil para un campo u otro. Por ejemplo, aquellos modelos con una resoluciónalta (si el parecido del modelo con la realidad es elevado), porque se ha encontrado un templatehomólogo cercano, puede ser muy útil en el estudio de la unión de ligandos, o en el diseño defármacos. Si la precisión del modelo es media (con un rango de 2.5 a 5 Å), porque hemos encontrado untemplate homólogo lejano, el modelo puede ser útil a la hora de localizar espacialmente residuosfuncionales importantes, o sitios activos, o sitios asociados a mutaciones.Y si el modelo tiene una baja resolución, porque pertenece a la categoría de Free Modeling, nospuede ser útil a la hora de identificar los límites de un dominio, en el reconocimiento topológico dela proteína o en su asignación a una familia o superfamilia [2].
Aproximación entre la precisión del modelo obtenido y la utilidad biológica de la estructura proteica.
6
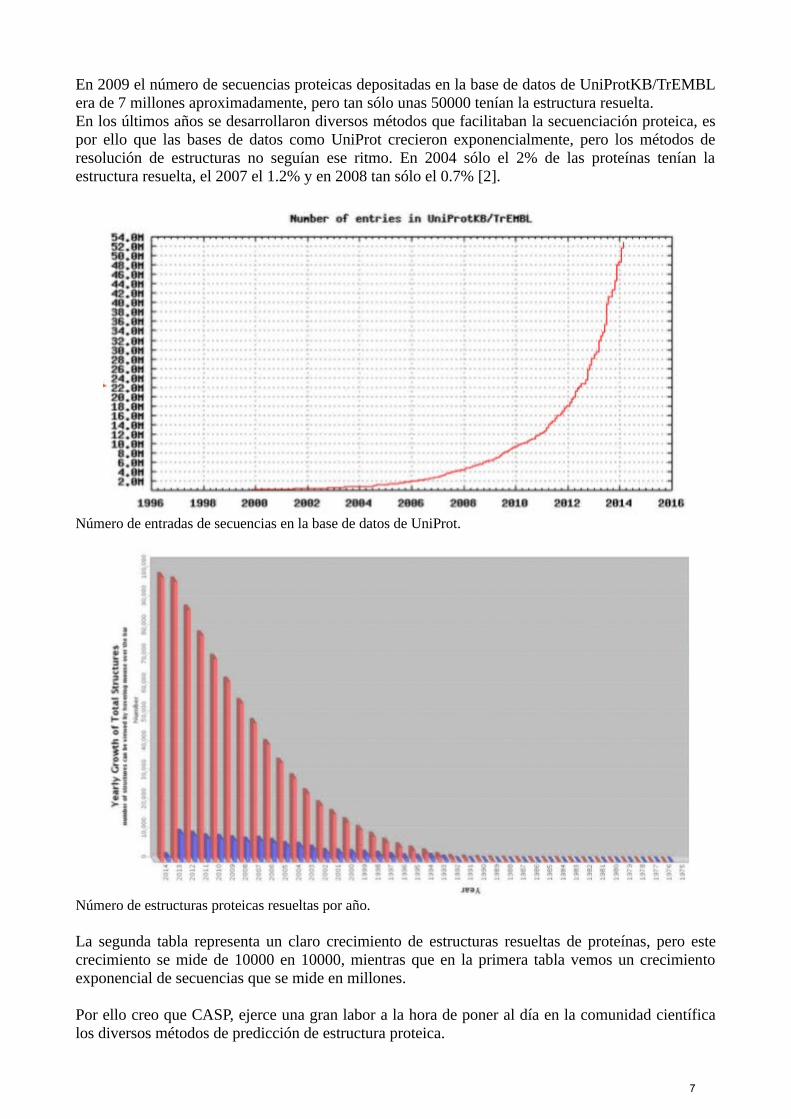
En 2009 el número de secuencias proteicas depositadas en la base de datos de UniProtKB/TrEMBLera de 7 millones aproximadamente, pero tan sólo unas 50000 tenían la estructura resuelta.En los últimos años se desarrollaron diversos métodos que facilitaban la secuenciación proteica, espor ello que las bases de datos como UniProt crecieron exponencialmente, pero los métodos deresolución de estructuras no seguían ese ritmo. En 2004 sólo el 2% de las proteínas tenían laestructura resuelta, el 2007 el 1.2% y en 2008 tan sólo el 0.7% [2].
Número de entradas de secuencias en la base de datos de UniProt.
Número de estructuras proteicas resueltas por año.
La segunda tabla representa un claro crecimiento de estructuras resueltas de proteínas, pero estecrecimiento se mide de 10000 en 10000, mientras que en la primera tabla vemos un crecimientoexponencial de secuencias que se mide en millones.
Por ello creo que CASP, ejerce una gran labor a la hora de poner al día en la comunidad científicalos diversos métodos de predicción de estructura proteica.
7
2.3 Estructuras limitadas
El número de plegamientos posibles no es ilimitado, si no que hay un número concreto deplegamientos, por lo que el número posible de posibilidades espaciales que tiene una estructuraproteica es limitado. De esto se deduce que aunque el número de secuencias conocidas hoy en díasea muy superior al número de estructuras resueltas, muchas de esas secuencias deben tener unaestructura muy semejante, es decir, que aquellas secuencias con un parecido razonable se plegaránde una manera similar.
Basándonos en esta información, nuestra labor principal durante el transcurso de CASP será la deencontrar una proteína similar a la nuestra (target), pero que ya tenga su estructura proteica resuelta(template). De esta manera alineamos ambas secuencias y generamos la estructura tridimensionalpara nuestro target.
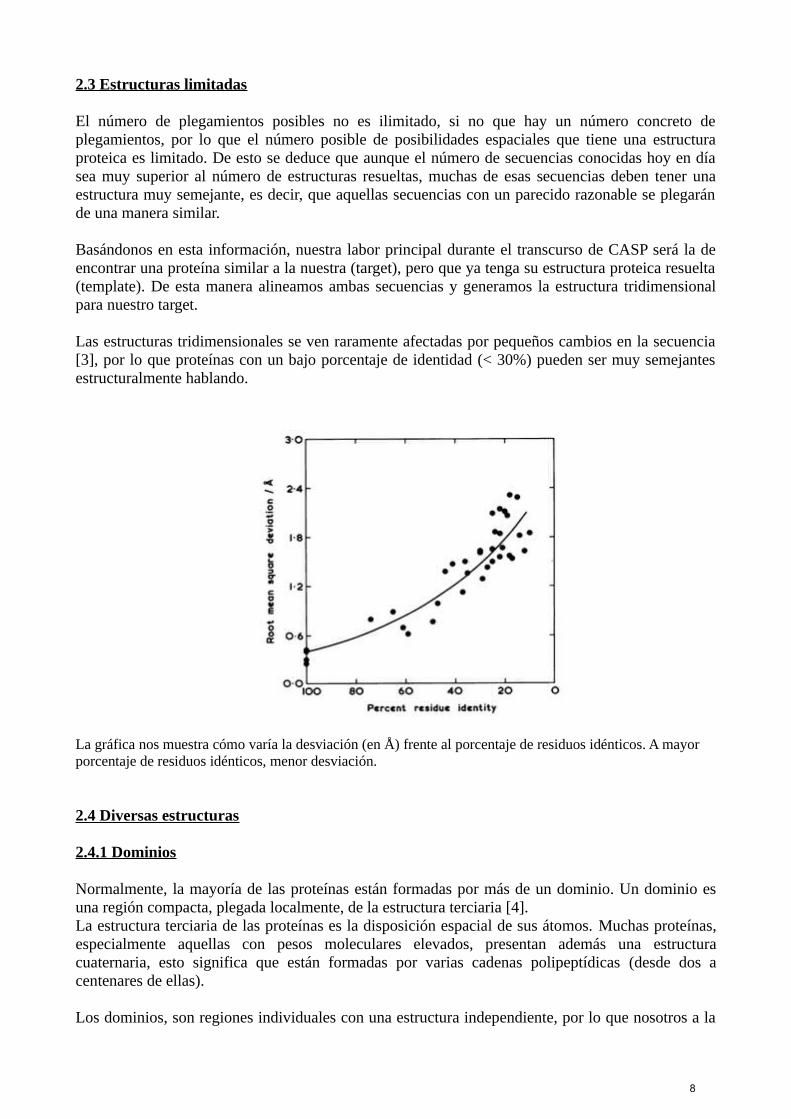
Las estructuras tridimensionales se ven raramente afectadas por pequeños cambios en la secuencia[3], por lo que proteínas con un bajo porcentaje de identidad (< 30%) pueden ser muy semejantesestructuralmente hablando.
La gráfica nos muestra cómo varía la desviación (en Å) frente al porcentaje de residuos idénticos. A mayor porcentaje de residuos idénticos, menor desviación.
2.4 Diversas estructuras
2.4.1 Dominios
Normalmente, la mayoría de las proteínas están formadas por más de un dominio. Un dominio esuna región compacta, plegada localmente, de la estructura terciaria [4].La estructura terciaria de las proteínas es la disposición espacial de sus átomos. Muchas proteínas,especialmente aquellas con pesos moleculares elevados, presentan además una estructuracuaternaria, esto significa que están formadas por varias cadenas polipeptídicas (desde dos acentenares de ellas).
Los dominios, son regiones individuales con una estructura independiente, por lo que nosotros a la
8

hora de predecir la estructura terciaria de la proteína, la dividíamos en dominios y predecíamos laestructura de los diferentes dominios de manera individual y luego tratábamos de encajar losdiversos dominios desde los que partíamos.
Los dominios están conectados entre sí mediante una cadena polipeptídica que suele tener algunaprolina que le otorga mayor movilidad. Estás uniones entre dominios las conocemos como links ysu longitud puede variar dependiendo de la proteína.Los distintos dominios suelen realizar funciones diferentes y en ocasiones un tipo determinado dedominio puede reconocerse en varias proteínas diferentes [4], lo que nos resulta realmente útil a lahora de predecir estructuras.
Una vez tenemos todos los dominios resueltos, procedemos a su ensamblaje haciendo uso deprogramas como Pymol o Modeller, principalmente. Programas de los que hablaré más adelante.Sobre la colocación de los diferentes dominios unos con otros no tenemos una idea exacta. Sabemosel orden que llevan, pues conocemos la secuencia proteica, pero su plegado con respecto a los otrosdominios no es muy claro. Lo que intentamos siempre es ensamblarlo de manera que la proteínaquede lo más compacta posible.Los asesores de CASP a la hora de evaluar las predicciones, también dividen la estructura final enlos diferentes dominios, por lo que el ensamblaje que nosotros realizamos no va a ser clave en laresolución, pero sí es verdad que un mayor plegado puede darnos unos pocos puntos de ventaja conrespecto a otros modelos.
Pero ¿cómo identificar los diferentes dominios de una proteína partiendo únicamente de lasecuencia proteica?.La verdad es que esto era realmente complicado.
Existen varios programas que nos dicen cuántos dominios hay en nuestra secuencia pero no sonpara nada exactos y lo más normal es que no se pongan de acuerdo. Pero podemos recabarinformación de diferentes apartados para obtener unas cuántas pistas. Podemos obtener informaciónde GeneSilico que es un metaservidor que recopila información de multitud servidores y nos es muyútil para hacernos una idea previa de la proteína con la que estamos trabajando. También usábamosHHpred, Pfam o incluso Firestar para determinar los dominios de nuestra proteína.
– En Genesilico – hay un apartado llamado “Protein Domains”, que recopila diversos métodosque nos indican el número de dominios de la proteína, pero no tiene nunca un acuerdoperfecto entre los diferentes servidores, excepto que la proteína sea muy pequeña queentonces predicen todos un único dominio. Nos sirve de guía pero poco más.
Esta es la proteína T0773, al ser una proteína de tan sólo 77 aminoácidos, la mayoría de servidores predicenun único dominio, exceptuando DoBo, que al ser una secuencia muy corta no es capaz de hacer supredicción.
9
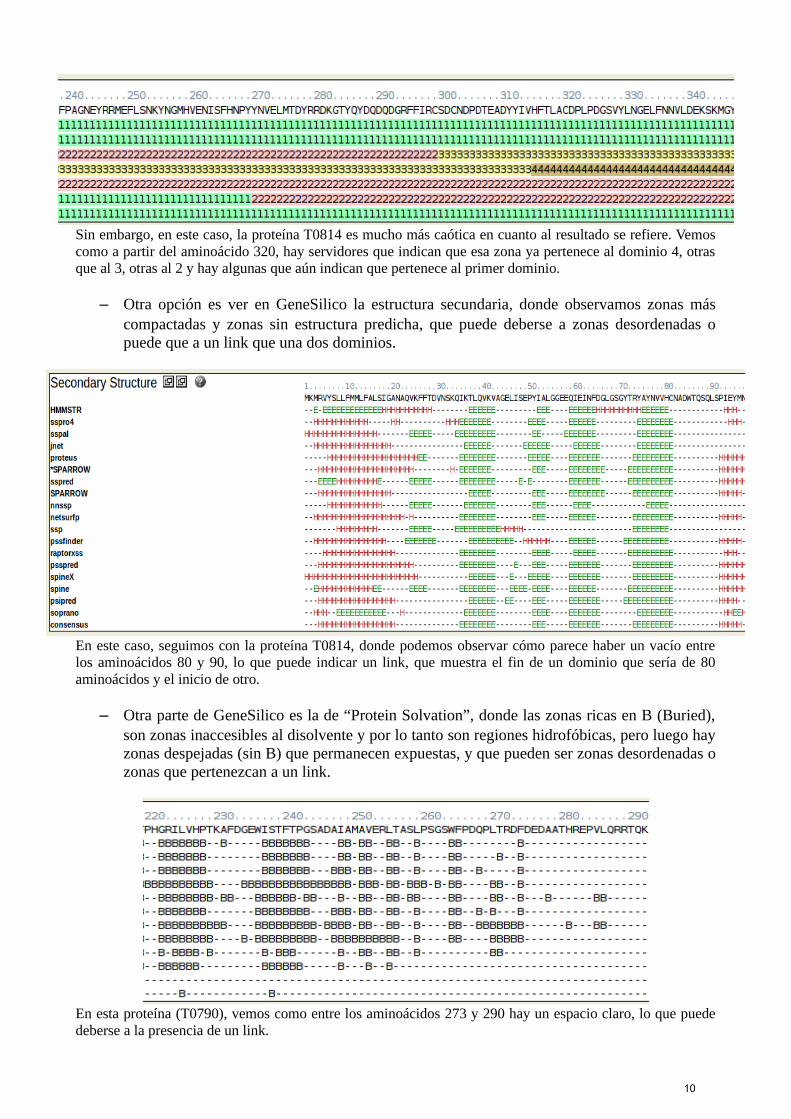
Sin embargo, en este caso, la proteína T0814 es mucho más caótica en cuanto al resultado se refiere. Vemoscomo a partir del aminoácido 320, hay servidores que indican que esa zona ya pertenece al dominio 4, otrasque al 3, otras al 2 y hay algunas que aún indican que pertenece al primer dominio.
– Otra opción es ver en GeneSilico la estructura secundaria, donde observamos zonas máscompactadas y zonas sin estructura predicha, que puede deberse a zonas desordenadas opuede que a un link que una dos dominios.
En este caso, seguimos con la proteína T0814, donde podemos observar cómo parece haber un vacío entrelos aminoácidos 80 y 90, lo que puede indicar un link, que muestra el fin de un dominio que sería de 80aminoácidos y el inicio de otro.
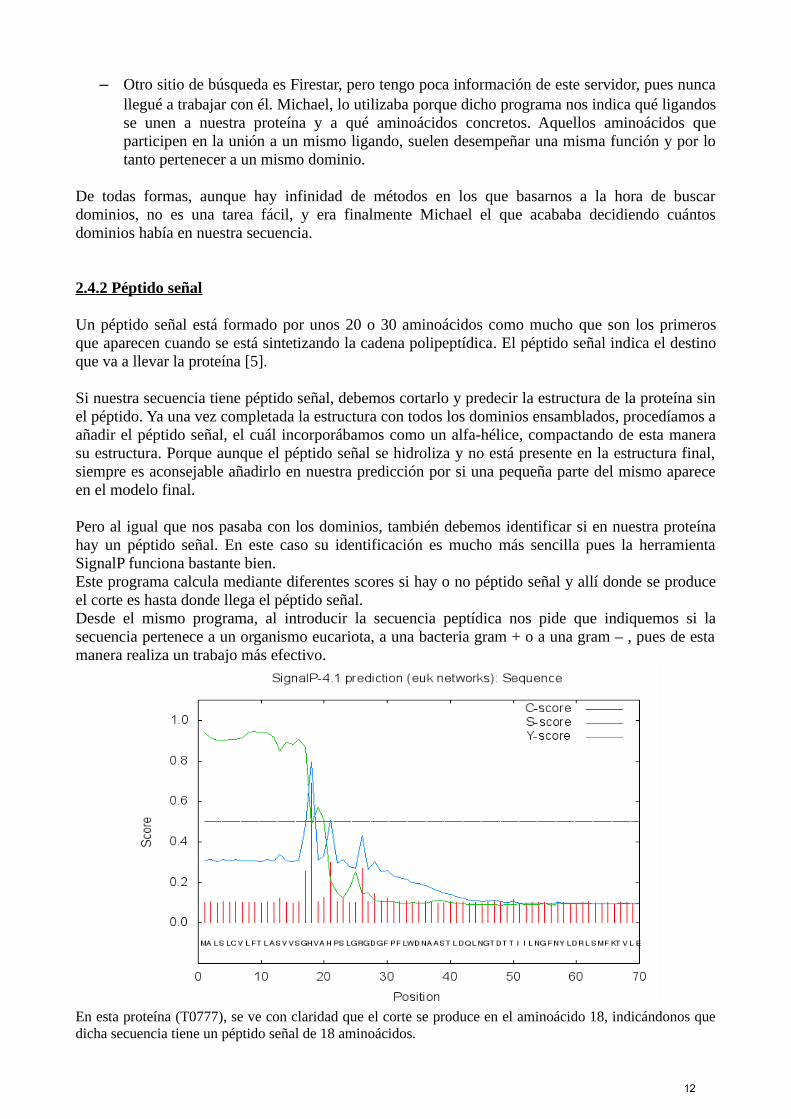
– Otra parte de GeneSilico es la de “Protein Solvation”, donde las zonas ricas en B (Buried),son zonas inaccesibles al disolvente y por lo tanto son regiones hidrofóbicas, pero luego hayzonas despejadas (sin B) que permanecen expuestas, y que pueden ser zonas desordenadas ozonas que pertenezcan a un link.
En esta proteína (T0790), vemos como entre los aminoácidos 273 y 290 hay un espacio claro, lo que puededeberse a la presencia de un link.
10

La accesibilidad al solvente puede determinar muchas propiedades como dónde se encuentran lossitios de unión al ligando, cómo interacciona la proteína con el medio, sitios funcionales, predicciónde superficies de interacción...La accesibilidad depende completamente de la hidrofobicidad de los aminoácidos además de estarsujeta a una gran restricción evolutiva, por las funciones tan importantes que desempeña.
– Otra pista la obtenemos con HHpred. Podemos ver que a lo mejor hay una parte queencuentra buenos templates (en rojo) y otra que no. Esa parte con buenos templates puedepertenecer a un dominio claro y el resto no se conoce a simple vista.
Esta es la proteína T0826 y mientras que en los 200 primeros aminoácidos no encontramos apenas nada, del213 en adelante se encuentran muchos templates buenos, lo que puede indicarnos que esa región correspondea un dominio.
– Otro sitio de búsqueda es en Pfam, donde se encuentran dominios y familias. Siencontramos información sobre un dominio podemos saber qué región ocupa en nuestrasecuencia.
Una vez que corremos Pfam, si se encuentra algún dominio, aparece de esta forma y debajo delgráfico nos muestra información sobre la familia del dominio, en qué aminoácido empieza y en cualacaba, el e-value, la longitud del dominio en aminoácidos …En este caso se encontró un dominio que pertenece a la familia CN_hydrolase en nuestra proteínaT0794.Hay otros casos en los que se encuentran más dominios
o incluso repeticiones de dominios intercalados
– Esto mismo que hemos hecho en Pfam, se puede obtener con Interpro
11
– Otro sitio de búsqueda es Firestar, pero tengo poca información de este servidor, pues nuncallegué a trabajar con él. Michael, lo utilizaba porque dicho programa nos indica qué ligandosse unen a nuestra proteína y a qué aminoácidos concretos. Aquellos aminoácidos queparticipen en la unión a un mismo ligando, suelen desempeñar una misma función y por lotanto pertenecer a un mismo dominio.
De todas formas, aunque hay infinidad de métodos en los que basarnos a la hora de buscardominios, no es una tarea fácil, y era finalmente Michael el que acababa decidiendo cuántosdominios había en nuestra secuencia.
2.4.2 Péptido señal
Un péptido señal está formado por unos 20 o 30 aminoácidos como mucho que son los primerosque aparecen cuando se está sintetizando la cadena polipeptídica. El péptido señal indica el destinoque va a llevar la proteína [5].
Si nuestra secuencia tiene péptido señal, debemos cortarlo y predecir la estructura de la proteína sinel péptido. Ya una vez completada la estructura con todos los dominios ensamblados, procedíamos aañadir el péptido señal, el cuál incorporábamos como un alfa-hélice, compactando de esta manerasu estructura. Porque aunque el péptido señal se hidroliza y no está presente en la estructura final,siempre es aconsejable añadirlo en nuestra predicción por si una pequeña parte del mismo apareceen el modelo final.
Pero al igual que nos pasaba con los dominios, también debemos identificar si en nuestra proteínahay un péptido señal. En este caso su identificación es mucho más sencilla pues la herramientaSignalP funciona bastante bien.Este programa calcula mediante diferentes scores si hay o no péptido señal y allí donde se produceel corte es hasta donde llega el péptido señal.Desde el mismo programa, al introducir la secuencia peptídica nos pide que indiquemos si lasecuencia pertenece a un organismo eucariota, a una bacteria gram + o a una gram – , pues de estamanera realiza un trabajo más efectivo.
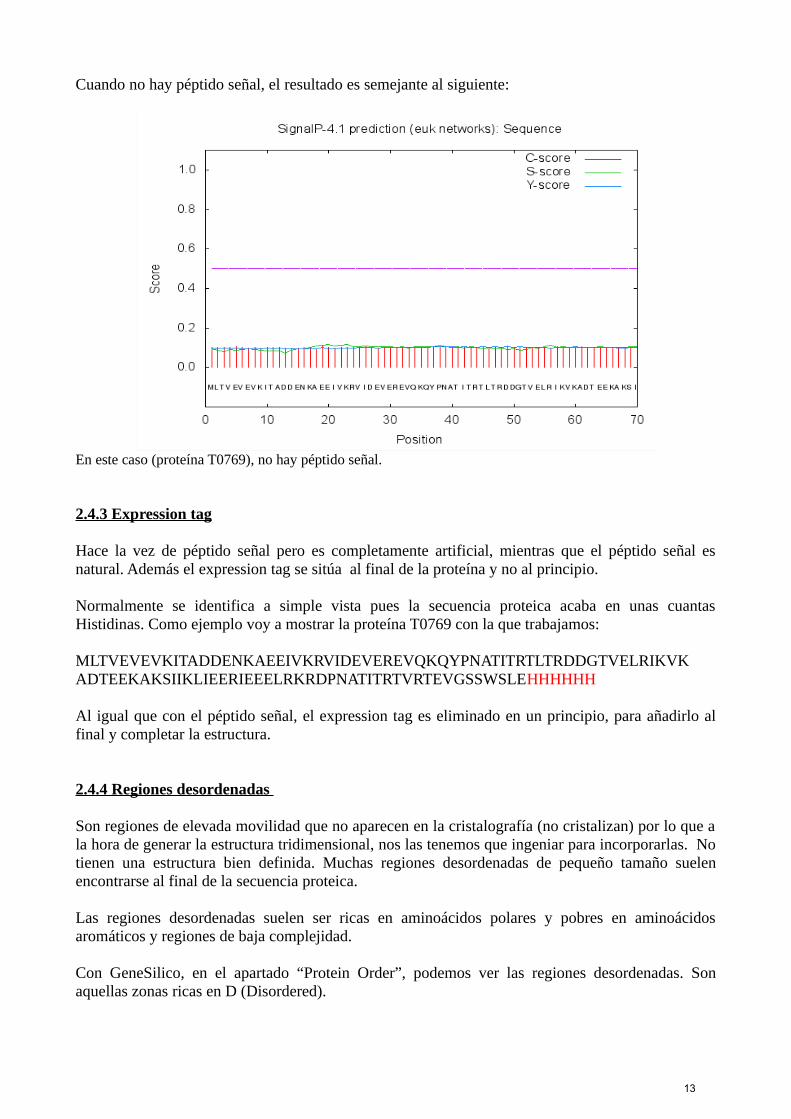
En esta proteína (T0777), se ve con claridad que el corte se produce en el aminoácido 18, indicándonos quedicha secuencia tiene un péptido señal de 18 aminoácidos.
12
Cuando no hay péptido señal, el resultado es semejante al siguiente:
En este caso (proteína T0769), no hay péptido señal.
2.4.3 Expression tag
Hace la vez de péptido señal pero es completamente artificial, mientras que el péptido señal esnatural. Además el expression tag se sitúa al final de la proteína y no al principio.
Normalmente se identifica a simple vista pues la secuencia proteica acaba en unas cuantasHistidinas. Como ejemplo voy a mostrar la proteína T0769 con la que trabajamos:
MLTVEVEVKITADDENKAEEIVKRVIDEVEREVQKQYPNATITRTLTRDDGTVELRIKVKADTEEKAKSIIKLIEERIEEELRKRDPNATITRTVRTEVGSSWSLEHHHHHH
Al igual que con el péptido señal, el expression tag es eliminado en un principio, para añadirlo alfinal y completar la estructura.
2.4.4 Regiones desordenadas
Son regiones de elevada movilidad que no aparecen en la cristalografía (no cristalizan) por lo que ala hora de generar la estructura tridimensional, nos las tenemos que ingeniar para incorporarlas. Notienen una estructura bien definida. Muchas regiones desordenadas de pequeño tamaño suelenencontrarse al final de la secuencia proteica.
Las regiones desordenadas suelen ser ricas en aminoácidos polares y pobres en aminoácidosaromáticos y regiones de baja complejidad.
Con GeneSilico, en el apartado “Protein Order”, podemos ver las regiones desordenadas. Sonaquellas zonas ricas en D (Disordered).
13

Aquí, en la proteína T0767, vemos como entre los aminoácidos 20 y 50 aproximadamente se ve una regiónrica en D, lo que nos está indicando que es una región desordenada.
2.4.5 Regiones transmembrana
A veces nos es muy útil saber si una proteína tiene parte de sus hélices-alfa atravesando lamembrana celular, pues nos puede ayudar a la hora de colocarlas al construir la estructuratridimensional de la proteína.
Esta es nuestra proteína T0830, que tiene 14 hélices transmembrana excepto aquellas que están en magenta,las cuales situamos por encima de las otras. Gracias al conocimiento de cuántas y qué hélices erantransmembrana, pudimos hacernos una mejor idea de la estructura.
Para predecir regiones transmembrana podemos mirar el apartado “TM Helices” de GeneSilico,donde aparecen unas “H” que representa las hélices transmembrana. Si hay símbolos negativos esque esa región de la secuencia se encuentra en la parte negativa de la membrana y si hay símbolospositivos, que se encuentra en la parte positiva. A veces, si hay un péptido señal, aparecerepresentado como “S”, al inicio de la secuencia, pero ya comenté que es mucho más fácil utilizar elprograma SignalP para este caso.
14
Y al igual que miramos SignalP para los péptidos señal, existe un programa equivalente para lashélices transmembrana conocido como TMHMM y nos da resultados como el siguiente:
Donde nos indican las regiones transmembrana y las regiones internas y externas a la membrana.Y en esta página, justo al lado de la gráfica hay una leyenda escrita que nos dice exactamente enqué aminoácidos empiezan y acaban las distintas regiones transmembrana.
2.4.6 Puentes disulfuro
El conocimiento de la presencia de puentes disulfuro en nuestra secuencia nos puede ser muy útil ala hora de construir la estructura tridimensional, pues generan unos plegamientos y acercamientosentre aminoácidos de manera que se imponen ciertas restricciones estructurales importantes.
Pero desgraciadamente en este caso no hay un programa que nos indique de manera clara si hay ono puentes disulfuro. Podemos basarnos en la información que nos aporta GeneSilico en el apartado“Disulfide Bridges”, que recopila la información de tres programas que buscan dichos puentes en lasecuencia. El resultado obtenido es semejante al siguiente:
15

Esta imagen pertenece a la proteína T0830, en la que vemos un puente disulfuro claro entre el aminoácido167 y el 192. De los tres programas, dos predicen el puente disulfuro representado por letras “a” minúsculaspor lo que estamos bastante seguros de la presencia del puente disulfuro, ya que los puentes disulfurosmarcados con la “a”, son más probables que los marcados con la “b”, y estos más que la “c”.
Aquellas proteínas propias de ambientes hostiles son ricas en puentes disulfuro al igual que lasproteínas extracelulares.
2.4.7 Coiled Coils
Las coiled coil o hélices superenrolladas son un motivo estructural de proteínas, en el que de 2 a 7hélices alfa se enrollan, siendo los dímeros y los trímeros las formas más comunes [6].Son estructuras características que nos interesa localizar, y la manera de conocer su existencia esvolviendo a trabajar con GeneSilico, fijándonos esta vez en el apartado “Coiled Coils”.Veremos algo semejante a esto:
Pertenece a la proteína T0832 la cual vemos que tiene predicción de coiled coil entre los aminoácidos 30 y55 aproximadamente.
2.4.8 Otras estructuras
A continuación voy a presentar unas pocas estructuras tridimensionales que he descubierto endiferentes targets durante mis meses de prácticas:
Kelch-type y Beta propeller - El dominio kelch se ve formado por un grupo de 5 a 7 repeticioneskelch las cuales forman una estructura terciaria conocida como beta-proppeller.Es decir, varios motivos kelch se unen para formar un beta-propeller.
16

Repetición rica en leucina (LRR) - es un motivo estructural de proteínas que forma un plegamientoen herradura [7][8] debido a la repetición de 20 a 30 aminoácidos de leucina.
La imagen pertenece al target T0827 y no tiene un aspecto de herradura perfecta pero sí se distingue la formamencionada. He coloreado las leucinas en magenta para que se pueda apreciar su elevada presencia en laestructura.
Histidina quinasa – Son proteínas típicamente transmembrana que juegan un papel importante en latransducción de señal a través de la membrana celular. La mayoría son homodímeros con actividadautoquinasa, fosfotranferasa o fosfatasa [9].
Repetición de tetratricopéptido (TPR) – es un motivo estructural. Consiste en un motivo de 34aminoácidos de la secuencia identificado en un alto número de proteínas. Se encuentra en grupos de3 a 16 motivos [10] los cuales forman scaffolds para mediar interacciones entre proteínas y amenudo ensamblar complejos multiproteicos.
Pertenece al target T0842 y creemos que puede ser un tipo de TPR.
Por último un dato que aprendí sobre los strands es que no aparecen por sí solos en la estructura, lohace siempre unido a otro strand como mínimo formando una hoja para estabilizarse. Por lo que sivemos sólo uno, es que esa estructura es parte de una estructura mayor (dímero, trímero …).
2.5 Vocabulario
Docking – El docking no es una estructura tridimensional pero es un término que llegamos a usarmucho con el que hacíamos referencia al acoplamiento molecular. Este es un método que predice laconformación preferida de un molécula, al estar unida a otra, con el fin de formar un complejoestable [11].
Sintenia genómica - Conservación del orden génico y la orientación a lo largo del cromosoma; unelevado grado de sintenia es indicativo de proximidad filogenética.
17

3. Material y Métodos
3.1 Bases de datos
A continuación presento varias de las bases de datos que hemos usado, en las que buscamos lassecuencias homólogas, los templates y cualquier información que nos pueda ser útil sobre nuestrotarget.
3.1.1 PDB
PDB es una base de datos que recoge todas las estructuras tridimensionales proteicas que seconocen. Es una herramienta muy útil para trabajar en CASP pues todas nuestras búsquedas detemplates se hacen en esta base de datos.
En PDB la estructuras resueltas están representadas por un código mezcla de letras y números y enocasiones llevan una letra extra que hace referencia a la cadena de la proteína.
Una vez introducido el código en la base de datos nos aparece a la derecha la apariencia de laestructura y en la zona central la información de la molécula en cuestión. Hay una pestaña con la que podemos ver la molécula en 3D pero lo que siempre hacíamos eradescargarla en formato .pdb y visualizarla en Pymol, un programa de visualización de estructurasmoleculares.
Otra pestaña es la llamada “Sequence” en la que podemos ver la información de SCOP, la de CATHy lo que nos resultaba más interesante, la estructura secundaria del template, la cual solíamoscomparar con la de nuestro target para ver los parecidos y diferencias y a qué se debían estas.
Ejemplo de visualización de la secuencia del template 1fk9.
3.1.2 SCOP y CATH
Tanto Scop como Cath son bases de datos de estructuras. Toman las estructuras de PDB y lasagrupan por su parecido estructural formando familias (de dominios semejantes), las cuales agrupanen superfamilias.Esto nos puede resultar útil a la hora de encontrar proteínas que tengan una baja identidad en sussecuencias aminoacídicas pero que se encuentren en una misma familia debido a que tienen unaestructura semejante, lo que puede facilitarnos la búsqueda de templates.
18

O también nos pueden indicar qué regiones se encuentran más conservadas dentro de una mismafamilia.
En CASP nos es muy útil esta información, por ejemplo, si tenemos un homólogo que no cubre todoel target, podemos mirar en SCOP o en CATH, qué ocurre en otras estructuras de su familia en laregión no cubierta, lo cual nos puede dar alguna pista.
SCOP y CATH aportan también una representación del universo estructural que conocemos. PDBestá muy sesgado hacia algunas estructuras que contienen un montón de estructuras.
Ambas bases de datos tienen diferencias conceptuales (por ejemplo, cómo se definen los niveles) ymetodológicas (SCOP es manual mientras que CATH es semiautomático). El grado de acuerdoentre ellas es, en general, alto aunque puede haber diferencias importantes en algunos casos.
Por ejemplo, cuando estamos trabajando con el metaservidor GeneSilico, si en el apartado deestructura secundaria vemos un mismo “Scop” para varias secuencias nos está indicando quepertenecen a la misma familia y que por lo tanto tienen un fold semejante.
3.1.3 PFAM
Pfam es una base de datos de dominios de las familias de proteínas que es ampliamente utilizadopor los biólogos para anotar y clasificar las proteínas. La base de datos consta de dos clases deentradas: Pfam-A, que contiene dominios de familias de proteínas bien caracterizadas con alineamientos dealta calidad, que se mantienen controladas manualmente.Pfam-B contiene secuencias de las familias que se han generado de forma automática mediante laaplicación del algoritmo Domainer [12].
Nosotros solíamos usar Pfam para encontrar los dominios de nuestro target. Si encontrábamos algúndominio, en Pfam se nos informaba de a qué familia pertenecía ese dominio.Esto nos resultaba muy útil porque si accedíamos a la familia aparecían varias opciones en esta basede datos. Lo mismo ocurría si accedíamos al CLAN o superfamilia.
Los diversos apartados nos indican:– Architecture – Nos muestra las diferentes arquitecturas en las que encontramos dicho
dominio, es decir, en qué posición se encuentra en la secuencia junto a otros dominios, o sipor el contrario lo encontramos de manera individual.
19

– Sequences – Alineamiento múltiple de las secuencias encontradas. En este caso procedíamosa su visualización con Jalview.
– Species – Representa la visualización de esa familia entre las especies.
Con Pfam también podíamos encontrar alineamientos para los diferentes dominios los cuales eranbastante buenos y por lo tanto útiles.
3.1.4 UniProt
UniProt (Universal Protein) – Es una base de datos de secuencias de proteínas e información de sufuncionalidad. Las dos fuentes principales de UniProt son Swiss-Prot y TrEMBL. En la primera, lassecuencias son anotadas a mano y revisadas y en la segunda las secuencias se anotan de maneraautomática y no están revisadas. Es por esto que en TrEMBL hay muchas más secuencias que enSwiss_Prot, pero siempre empezamos con Swiss_Prot pues su información es de mucha máscalidad.
3.2 Clasificación de métodos de predicción
El modelado de una secuencia basado en estructuras tridimensionales ya conocidas se resume encuatro importantes pasos: encontrar los templates, alinear nuestra secuencia con la del template,construir el modelo y evaluarlo [13].
Los templates para el modelado suelen encontrarse por métodos de comparación de secuencias,como BLAST o PSI-BLAST [14], o por métodos de threading [15], de los que hablaré másadelante.
La precisión o exactitud de un modelo está relacionada con el porcentaje de identidad de nuestrasecuencia con la del template [16], a mayor identidad, mayor exactitud del modelo.Los modelos con una elevada exactitud son aquellos con una identidad superior al 50% a la de sustemplates. Los modelos con una precisión media son aquellos con una identidad que oscila entre el30 y el 50%. Y por último aquellos modelos menos precisos son debidos a que tienen un porcentajede identidad menor al 30% [16].
En nuestro caso, durante todas las prácticas con CASP, rara vez encontramos templates con unaidentidad superior al 30%.
3.3 Métodos de Predicción
Homología cercana – Si encontramos un template con una identidad de secuencia superior al 30%,lo normal es que el modelo que obtengamos sea bueno pues hemos encontrado un templatehomólogo. Los modelos en este caso entran dentro del grupo de exactitud media o alta y se consideran TBM.
Homología remota – Si la identidad oscila entre el 20 y el 30%, hablamos de homología remota,donde los modelos se consideran poco precisos pero nos podemos acercar al original con un buentrabajo. Aunque sea remoto, si encontramos templates para el modelo este se sigue considerandoTBM.
Podemos distinguir dos tipos:
20

– Basado en secuencia: Tratamos de buscar secuencias homólogas remotas mediante búsquedade perfil contar perfil. Estas secuencias pueden haber sufrido numerosos cambios en losresiduos pero los más importantes y con función biológica clara se mantienen.Para realizar estas búsquedas usamos herramientas como HHpred (que es una mezcla de losmotores de búsqueda HHblits y HHsearch) o HMMER.
– Basado en secuencia y estructura (Fold Recognition): Las técnicas de fold recognitionencuentran templates que los métodos basados en secuencia no pueden porque usaninformación estructural así como similitud de la secuencia para encontrar templates.Los programas de fold recognition usan una base de datos de plegamientos donde sealmacenan todas las estructuras tridimensionales proteicas conocidas e intentan encajar eltarget en todas esas estructuras (alineando secuencia a estructura) dando un score a cada unoy eligiendo el mas apropiado.A este proceso también se le conoce como threading. Y la verdad funciona bastante bien,pues se pueden encontrar homólogos muy lejanos con los que sería muy difícil dar con ellossi se busca únicamente mediante secuencias.Una gran parte de los servidores trabaja con técnicas de Fold Recognition.
Ab initio - Para este caso no tenemos template en el que basarnos a la hora de predecir nuestraestructura. Estos métodos requieren un vasto poder computacional y unos altos conocimientosfisicoquímicos porque no tienen información alguna sobre estructuras conocidas.Los métodos ab initio trabajan bien con proteínas pequeñas pero cometen muchos errores conproteínas medianas y grandes.
De Novo – Es semejante a los métodos ab initio pero en este caso se usan fragmentos de proteínascomo modelo. Aun así no podemos afirmar que use templates pues son sólo fragmentos.Funciona mejor que los métodos ab initio pero sigue siendo únicamente útil en proteínas pequeñas.La exactitud y fiabilidad de los modelos producidos por métodos de Novo, son mucho más bajasque los modelos basados en alineamientos con más de un 30% de identidad de secuencia, pero latopología básica de la proteína o de un dominio en concreto pueden ser predichos razonablementebien [15].
Servidores como ROBETTA realiza modelado proteico ab initio, aunque también puede modelarbasándose en templates. Por otro lado el servidor QUARK funciona bastante bien en el modeladode Novo.
Los modelos de baja resolución producidos por estos métodos (ab initio y de novo) pueden revelarrelaciones estructurales y funcionales entre proteínas que no parecían emparentadas basándonos ensus secuencias aminoacídicas y también pueden proporcionar un marco de estudio que permitaanalizar las relaciones espaciales entre residuos evolutivamente conservados o entre residuos quemuestren una funcionalidad importante [15].
Tanto los métodos ab initio como los de novo son usados para FM.
3.4 Búsqueda de secuencias
Principales servidores que empleamos para la búsqueda de secuencias:
21

3.4.1 BLAST
Es una herramienta muy útil que usé con todos los targets. Primero usaba como base de datos “nr”(Non redundant protein sequences), lo que me servía para adquirir información sobre mi secuenciaproteica, como a qué familia y a qué organismo pertenecía; el número de secuencias semejantes quepodía encontrar; si mi secuencia era un fragmento de una secuencia mayor, lo que nos puederesultar muy interesante a la hora de buscar homólogos lejanos ya que podemos encontrarinformación que no encontraríamos o nos sería más costoso si buscásemos sólo con nuestro targeten lugar de con la secuencia entera.Un ejemplo explicativo nos ocurrió con el target T0837. Al ampliar el fragmento vimos que la partede la secuencia que no aparecía era una región transmembrana, lo que influyó posteriormente ennuestra predicción.
El aspecto de una búsqueda con BLAST en la base de datos nr es el siguiente:
Pertenece al target T0767 y en la parte superior podemos ver que nos indican la superfamilia y en la gráficainferior el score del alineamiento.
Posteriormente a esta búsqueda procedía a usar de nuevo el BLAST, pero esta vez contra la base dedatos de PDB, por si podíamos encontrar algún template interesante, pero muy rara vezencontrábamos algo.
22

Con el target T0767 fue de los pocos con los que encontramos información. La línea roja equivale altemplate 4QPV_A del PDB con una identidad del 98%.
3.4.2 PsiBLAST
Después de llevar a cabo mis búsquedas con BLAST, si no encontraba nada contra la base de datosde PDB, que solía ser la mayoría de veces, realizaba un PsiBLAST contra PDB, ya que PsiBLASTes un proceso iterativo que compara perfiles contra la base de datos, de esta manera podemosencontrar más fácilmente homólogos remotos.He de decir que los resultados que obteníamos con PsiBLAST no eran tampoco muy abundantes,pues muchas veces no encontrábamos nada, pero sí eran más numerosos que con BLAST. Esto esbastante evidente pues la mayoría de los targets que tuvimos que analizar son targets complicadosque o no tienen template o los que tienen son difíciles de encontrar.
3.4.3 Jackhmmer
Jackhmmer es el método de búsqueda iterativa del paquete HMMER y es análogo a PSI_BLAST.Una búsqueda típica en Jackhmmer comienza con nuestra secuencia la cual es buscada frente a unabase de datos de proteínas como UniProt o NR, convirtiendo así la secuencia en un perfil HMM.Las secuencias que superen el umbral para esta primera búsqueda serán alineadas y usadas paraconstruir un segundo perfil HMM. Este segundo perfil se busca frente a la misma base de datosanterior encontrando homólogos más distantes que los encontrados en la primera búsqueda. Y deesta manera se continúa con las siguientes iteraciones.
Nosotros solíamos hacer entre dos y cuatro iteraciones, pues tampoco es conveniente alejarsemucho de nuestra secuencia original.
En un primer vistazo de la búsqueda en Jackhmmer con el target T0838, podemos distinguir alinicio que en este servidor también encontramos información del número de dominios que nosaporta PFAM y de si hay regiones transmembrana o péptido señal.
Y más abajo encontramos las diferentes secuencia homólogas encontradas.
23

Al realizar una segunda iteración se nos informa del número de nuevas secuencias que han sidoañadidas.
Si nos desplazamos a la parte inferior de la página vemos una gráfica que nos muestra losaminoácidos que más se repiten en cada zona de la secuencia. Es muy útil para observar quéaminoácidos se conservan y cuales varían.
En Jackhmmer en el apartado “Domains” podemos visualizar los dominios y además su posición entodas las cadenas.
En la pestaña “Downloads” nos podemos descargar el alineamiento múltiple en varios formatos.Nosotros nos lo descargábamos en formato FASTA para visualizarlo en Jalview.
Podemos también ver la información de los resultados de Jackhmmer en Uniprot, que si es “review”tiene información contrastada y de calidad.
3.4.4 HHBlits
Otra forma de buscar secuencias es con el motor de búsqueda Hhblits de Hhpred.HHBlits es un motor de búsqueda que trabaja con perfiles HMM que compara con su base de datosy en comparación con PSI-BLAST, HHblits trabaja más rápido, tiene un 50-100% mayorsensibilidad y genera alineamientos más precisos [17].
Tanto HHblits (base de datos de perfiles) como HHMER (base de datos de secuencias), realizanbúsquedas de perfil contra perfil, llevando a cabo una búsqueda más detallada lo que nos permitíalocalizar homólogos remotos.Trabajando con HHblits y con HHMER realizábamos ocho grupos diferentes de búsqueda desecuencias, cuatro con HHblits y cuatro con HHMER. En cada grupo de cuatro variábamos el e-value con el que buscábamos, buscando primero con un e-value de 10-40, luego con uno de 10-10,otro de 10-4 y por último con un e-value de 100. Con -40 era muy restrictivo y apenas obteníamosresultados y con 0 lo usábamos cuando no encontrábamos apenas nada con el resto de e-values. Losmás empleados por lo tanto eran -10 y -4.
3.4.5 Número de secuencias
Al principio pensaba que cuantas más secuencias encontrásemos, mejor sería a la hora de generarlos alineamientos. Pero en realidad la mejor opción se encuentra en el término medio. Si tenemosmuy pocas secuencias es más difícil observar si una región se conserva o no, pues tenemos menossecuencias con las que comparar. Pero tener muchas secuencias no siempre es bueno, pues puedencarecer de variabilidad o darnos un exceso de información que nos desvíe de nuestra búsquedadando errores en los alineamientos.Lo mejor entonces es tener un número elevado sin ser excesivo de secuencias y que estas seanvariables, pero desgraciadamente esto no es lo que solíamos encontrar.
3.5 Búsqueda de templates
HHpred
Detecta proteínas homólogas (templates) y predice estructuras por comparación HMM-HMM.
24

Empezando con nuestra secuencia de inicio, HHpred construye un alineamiento con múltiplessecuencias usando HHblits y lo transforma en un perfil HMM. Esto se compara con una base dedatos de HMMs que representa proteínas con estructura tridimensional conocida (ej. PDB, SCOP) ofamilias de proteínas anotadas (ej. PFAM, SMART, CDD, COGs, KOGs). En HHpred además de nuestro target, también podemos enviar alineamientos o incluso perfiles, deesta manera ampliamos el rango de búsqueda.La información que se nos muestra es una lista de homólogos cercanos y sus alineamientos connuestra secuencia de inicio.
Esta lista muestra los homólogos cercanos para el target T0826
Podemos destacar las siguientes columnas:En la primera columna encontramos el nombre e información de los diferentes templates.Prob – probabilidad del template de ser un verdadero positivo.E-value – da el número promedio de falsos positivos.P-value – equivale al E-value dividido entre el número de secuencias en la base de datos.Query HMM – región de nuestra secuencia que alinea con el template.Template HMM – región del template que alinea con nuestra secuencia.
Aquí vemos el inicio del alineamiento entre nuestra secuencia y el primer template.
Los colores representan diferentes categorías de los residuos, es decir, un mismo color indica quelos residuos son similares en cuanto a sus características (por ejemplo: hidrófobos, polares ...)
A simple vista debemos fijarnos en varios detalles, por ejemplo en la similitud y en la identidad. Eneste caso la identidad es del 15%, la cual es baja pero la mayoría de casos fueron así.
25

La similitud es útil conocerla cuando la identidad es baja, ya que si aun siendo baja la identidad, lasimilitud es buena, el template puede sernos útil.
Debemos fijarnos en los perfiles, que son las líneas que pone “Consensus”. Hay un perfil paranuestra secuencia (Q Consensus) y otro para el template (T Consensus). Siempre miramos si estosperfiles son buenos o si por el contrario son un desastre y no nos sirve el alineamiento para nada enabsoluto. Podemos distinguir dos tipos de perfiles sin utilidad: uno, que es evidente, se produce cuando no separecen en nada los perfiles de la secuencia original y del template; y el otro sucede cuando ambosperfiles son idénticos, lo que nos está indicando que directamente no hay perfil.
Ambos perfiles se relacionan mediante diferentes símbolos que indican el nivel de adecuación entreambos. Su significado es el siguiente:| → Muy bueno + → Bueno . → Neutro - → Malo = → Muy malo
Las líneas Q ss_pred y T ss_pred indican la predicción de estructura secundaria según PSIPRED.Podemos encontrar tanto letras mayúsculas como minúsculas. Las mayúsculas indican que es másprobable encontrar ese tipo de estructura, por lo tanto es más fiable.
La línea T ss-dssp, informa sobre la estructura real del template calculada con el analizador DSSP.Este toma los datos de PDB, y a cada aminoácido de la secuencia le asigna un punto y un valor de latabla de ángulos, dándole una letra que indica a qué estructura puede pertenecer. Se basa enpatrones que tienen en cuenta la geometría y los puentes de hidrógeno.
T = giro estabilizado por puentes de HidrógenoH = alfa-hélice, ~4 aa por vueltaG = hélice 310, ~3 aa por vueltaI = hélice phi, ~5 aa por vueltaB = conformación betaE = conformación beta formando láminaS = giro (sin puentes de Hidrógeno) [18]
Este sistema no predice, sólo anota e interpreta los datos ya existentes.
Los gaps o espacios que aparecen en los alineamientos son zonas que no alinean. Aquellosalineamientos con multitud de gaps son nefastos y de los que se puede sacar muy poca información.
Tras toda esta información, en el ejemplo que he puesto, tanto la identidad como la similitud sonbajas pero hay regiones muy buenas que se pueden aprovechar a la hora de hacer la predicción. Hayzonas interesantes con varios (+) y una zona muy concreta que abarca una lámina beta que es ricaen (|).
Por último, añadir que HHpred también puede predecir estructuras tridimensionales de proteínashaciendo uso de los templates homólogos que encuentra en PDB. Para crear estos modelos trabajajunto al programa Modeller.A veces hemos empleado los modelos que HHpred generaba pero los alineamientos nunca eran deltodo buenos por lo que preferíamos mejorarlos manualmente y aplicar nuestro alineamientodirectamente en Modeller.
26

3.5.1 Si no hay template o es remoto
Cuando no encontrábamos un template con PsiBlast o con Hhpred, antes de tachar el modelo comoFree Modeling realizábamos una búsqueda más exhaustiva con el fin de encontrar un templateremoto. Para ello contábamos con la colaboración de Federico Abascal que mediante diversastécnicas nos ayudaba a encontrar secuencias homólogas de difícil localización porque a lo mejor suse-values no eran muy significativos.
Una de las técnicas era investigar si la secuencia con la que estamos trabajando (nuestro target)pertenece a una secuencia mayor. Si es así, amplía la búsqueda usando como referencia la secuenciamayor, y en cada búsqueda elimina las secuencias redundantes que no aportan información y así vaencontrando homólogos a esa secuencia y se queda con los homólogos que comparten zona connuestro target. Esos homólogos son remotos, pero pueden resultarnos muy útiles aunque sólocompartan una región del target.
Otra de las técnicas que manejaba Federico era la de generar alineamientos para el target yalineamientos para los homólogos encontrados y luego estudiaba si estos alineamientos coincidían yen qué partes lo hacían, es decir, que residuos se mantenían.
Y por último estudiaba las regiones conservadas, las familias (haciendo mucho uso de Pfam) ycomparaba información de varios papers; en definitiva fue de gran ayuda en la creación de modelosbasados en templates remotos.
Por el contrario si no había template alguno nos enfrentábamos a un FM y para trabajar con él nosbasábamos en los modelos generados por los diversos servidores que participaban en CASP.
3.6 Alineamientos
La secuencia de una proteína determina qué estructura tridimensional tendrá esta, es por ello querealizamos alineamientos con secuencias homólogas para estudiar los cambios y las zonasconservadas. Dos secuencias son homólogas si tienen un origen común. No hay niveles niporcentajes de homología, dos secuencias son homólogas o no lo son.Si alineamos únicamente dos secuencias, hablamos de un pairwise, y si alineamos varias, estamoshablando de un alineamiento múltiple. Los alineamiento múltiples de secuencias es un métodofundamental para la comprensión de la función de los grupos de secuencias de ácidos nucleicos ysecuencias de proteínas [19].
Los cambios que podamos encontrar entre secuencias de proteínas homólogas pueden deberse a queesas proteínas, aunque tengan un origen evolutivo común, pueden que realicen ahora diferentesfunciones o que haya variado el modo de vida del organismo, lo que puede requerir diversoscambios en la estructura tridimensional.
Pero nosotros a la hora de comparar secuencias, lo que realmente nos interesa son las zonas másconservadas, aquellas regiones que al ser fundamentales para la proteína, no han variado con eltiempo. Y en aquellas zonas conservadas, las estructuras tridimensionales se mantienen, por lo queencontrar estas zonas nos es de gran ayuda a la hora de predecir la estructura.
En muchas ocasiones en los alineamientos, los aminoácidos no son exactamente los mismos, peroson aminoácidos con propiedades muy semejantes, por lo que debemos considerarlos como regiónconservada.
27

Un problema que solemos tener a la hora de buscar secuencias homólogas es encontrar secuenciasque son parecidas por azar pero que no nos son útiles pues no tiene ninguna homología con nuestrasecuencia. Por ejemplo alguna secuencia que hemos manejado era rica en leucinas, por lo que otrasecuencia rica en leucinas podría aparecer como homóloga aunque no lo fuese. Para descartar estassecuencias debemos fijarnos en el e-value, el cual depende del tamaño de la base de datos pero conun valor inferior a 1e-0.5 nos podemos fiar. El problema venía cuando había homólogos remotoscon un e-value muy poco significativo pero que nos podía interesar.
Nosotros empleábamos HHBlits y HHMer para realizar los alineamientos entre secuencias.Alineábamos principalmente los ocho grupos de secuencias por separado (ocho alineamientosdiferentes) que antes he comentado. Esos alineamientos podían ser visualizados en estos programaso usando JalView.
3.6.1 Jalview
Jalview es un programa donde podemos visualizar de manera sencilla y práctica los alineamientosmúltiples. Es además un editor pues tiene numerosas herramientas con las que manejar losalineamientos.
Este es el aspecto que puede presentar un alineamiento múltiple en Jalview.
Los aminoácidos teñidos con el mismo color son aquellos con características semejantes, por lo queaquellas regiones de un mismo color son regiones conservadas.
Con este programa podemos visualizar las partes hidrofóbicas de las secuencias, las que semantienen constantes, las partes que tienden a formar hélice, las que tienden a formar strand …
Todos estos programas eran empleados cuando alineábamos múltiples secuencias para detectar laregiones conservadas y predecir los contactos que ahora explicaré. Sin embargo a la hora de alinearnuestro target con el template, (era un alineamiento normalmente de dos o tres secuencias, el targetmás los posibles templates), al tratarse de un alineamiento del que dependía nuestro modelo, eraMichael el que lo hacía a mano.
28

3.7 Predicción de Contactos
Entendemos como contacto al acercamiento entre residuos de la estructura. Aquellas distanciasinferiores al umbral de 8 angstroms son consideradas como contactos [20].El poder predictivo de los contactos puede resultarnos muy útil a la hora de predecir modelostridimensionales de proteínas ya que los contactos existentes dentro de una proteína generanimportantes restricciones estructurales que debemos tener en cuenta si queremos generar un buenmodelo tridimensional.
Los contactos nos pueden resultar útiles tanto a la hora de generar el modelo como a la hora deevaluar los diferentes modelos obtenidos.En teoría las predicciones de contactos a más de 24 residuos de distancia son mejores a la hora deevaluar restricciones en una estructura tridimensional [20], pues aquellos contactos que se producenentre residuos cercanos no afectan en gran medida a la estructura proteica. Aun así es convenientetenerlos en cuenta pues pueden aportarnos información útil.
También es conveniente destacar, que la predicción de contactos no es igualmente aplicable enproteínas globulares que fibrosas. Las proteínas globulares reciben este nombre debido a que suscadenas polipeptídicas se pliegan en estructuras compactas muy distintas de las formas filamentosasy extendidas de las proteínas fibrosas [4].El problema surge con las proteínas fibrosas, ya que si se encuentran muy extendidas, apenas haycontactos con los que poder trabajar.
Podemos obtener contactos de dos formas diferentes:
Coevolución
También conocido como mutaciones correlacionadas. Es una predicción que se basa en la búsquedade covariación entre posiciones en un alineamiento múltiple. Este comportamiento puede deberse ala cercanía espacial ya que si ocurre una mutación en una zona de la proteína, esta sólo es viable siocurre una segunda mutación en un residuo cercano que compense la primera. Es decir, si dosresiduos hacen contacto en la proteína y uno de ellos muta, el otro residuo se ve favorecidoevolutivamente a mutar para que se siga manteniendo el contacto.
Los predictores de contactos por coevolución que usamos durante las prácticas fueron cuatro:simone, pcons, plmdca y psicov:
Psicov es el mejor de todos cuando hay pocas secuencias pero funciona peor cuando hay muchas. Pcons suele estudiar los contactos entre residuos cercanos, al contrario que psicov, que suelesepararlos más. Plmdca es el mejor cuando hay muchas secuencias.Simone es el predictor creado por nuestro grupo y es la primera vez que se emplea.
El principal problema que surge a la hora de usar este tipo de predictores por coevolución es que siel número de secuencias encontradas es muy escaso, no vamos a obtener ningún tipo deinformación, pues nos es imposible generar alineamientos en los que encontrar coevolución.Además las secuencias que empleemos deben tener diversidad para que se produzcan lasvariaciones que buscamos y que estas además sean significativas.
En este tipo de predicciones podemos encontrar una sobrepredicción de contactos en los laterales dela proteína, pues estas zonas equivalen a regiones menos conservadas de la proteína y por lo tantomás variables.
29

Consenso
Los predictores de contactos por consenso son también llamados Model Contacts.En este caso, lo primero que hacemos es fijarnos en los contactos que más se repiten en todos losmodelos generados por los servidores que participan en CASP. Esos son lo que podemos considerarcomo contactos fiables. Para obtener estos contactos, se calcula la distancia en angstroms entre cada par de carbonos alfa decada modelo. Nos fijaremos entonces en aquellos modelos que tengan un mayor número de contactos fiables,pues serán más semejantes al modelo que queramos predecir.
Si la mayoría de los modelos generados por los servidores son muy semejantes, nos van a aportarmuy poca información ya que no vamos a poder obtener los contactos que son más comunes a todoslos modelos. Es decir, si la variedad entre los modelos es escasa, nos van a aparecer los mismoscontactos en todos. Si por el contrario los modelos son muy diferentes entre sí, tampoco nos va aservir de mucha ayuda, pues apenas se encontrarán contactos comunes entre ellos.
En este caso el método de predicción de contactos por consenso siempre es aplicable, pues tan sólonecesita de los modelos generados por los servidores los cuales siempre están disponibles.
Nosotros a la hora de trabajar con los contactos entre residuos no nos decantábamos en un principiopor un método u otro. Si no obteníamos secuencias estaba claro que dependíamos únicamente de lapredicción de contactos por consenso, pero si teníamos un número suficiente de secuencias buenas,comparábamos resultados entre ambos métodos hasta obtener el resultado que más nos satisfacía.
3.7.1 L-factor
El L_factor es un método con el que se determina cuantas predicciones de contactos se toman enfunción del tamaño de la proteína. Debemos tener en cuenta que cuanto mayor sea una proteína máscontactos va a haber en proporción a su tamaño.
3.8 Métodos de predicción de Estructura Secundaria
Los programas más empleados a la hora de estudiar la estructura secundaria eran:
3.8.1 PSIPRED
PSIPRED (Psi-blast based secondary structure prediction) es uno de los mejores predictores deestructura secundaria.
30

Este ejemplo pertenece a los primeros 120 aa del target T0806.
La fiabilidad del resultado es buena basándonos en la línea Conf (confidence of prediction) ypredice un strand al inicio y 5 hélices.
3.8.2 JPRED
JPred predice la accesibilidad del solvente y las regiones Coiled-Coils con el método Lupas y laestructura secundaria con el algoritmo Jnet [21].
Análisis de JPRED con el target T0806.
En lo que más nos fijábamos era en la estructura secundaria predicha, donde aparecen las hélices enrojo y los strands en amarillo; y en el indicativo de números de la parte inferior que nos indicancuan fiables son los resultados. Cuanto mayor sea el número más fiable es el resultado.Por ejemplo en este caso aparece un strand al final que está señalado con varios 0 lo que nosadvierte de que no es nada seguro que allí haya una lámina beta. Es más, en PsiPred no apareceningún strand en esa posición, por lo que todo parece indicar que no hay strand alguno en esaposición.
Para el resto de aminoácidos ambos predictores parecen mostrar unos resultados muy semejantes.Esto nos facilitaba mucho las cosas a la hora de decidirnos en nuestra predicción.
3.8.3 GeneSilico
GeneSilico es un metaservidor con gran número de apartados que empleábamos para obtener unainformación completa de nuestro target, no sólo para la estructura secundaria. Corríamos elprograma tanto con el target entero como con el target dividido en sus diferentes dominios si es quelos tenía.
En GeneSilico el usuario puede enviar una secuencia de aminoácidos o una alineación desecuencias múltiples a un conjunto de métodos para la predicción de la estructura primaria,secundaria y terciaria [22].
Los servidores de GeneSilico son los siguientes:– Estructura primaria – hmmpfam y hhsearch_cdd
31

En el que me solía fijar era en hmmpfam que me podía dar información sobre los posibles dominiosdel target, aunque en este caso era mucho más fiable ver los resultados en la pagina web de PFAMdirectamente.
– Estructura secundaria – este apartado tiene muchos subgrupos de los cuales ya he hablado deellos anteriormente. Son los siguientes: Protein Domains, Disulfide Bridges, TM Helices,Protein Order, Coiled Coils, Protein-DNA interaction (nos indica qué zonas del de laproteína interactúan con el DNA), Protein-RNA interaction (qué zonas interactúan conRNA), Protein Solvation y Secondary Structure.Este último apartado concentra una gran cantidad de servidores por lo que es complicado que lleguen a un consenso, pero siempre nos sirve de ayuda. Para concretar más en la estructura secundaria usábamos más los programas antes mencionados (PSIPRED y JPRED).
- Estructura terciaria – Están los siguientes servidores: pdbblast, csblast, coma, hhblits, prc, blastp, ffas, hhsearch, mgenthreader y sp3.Entre estos servidores, si el target no es sencillo solía haber mucha discrepancia entre servidores y muchos gaps, lo que solía ser lo habitual.Los colores de las secuencias de estos apartados de estructura terciaria deben coincidir con los de la estructura secundaria. Es decir, deben coincidir las estructuras predichas en la estructura secundaria con los de la terciaria.
A la hora de interpretar los resultados de la estructura terciaria nos fijamos en el score, y también enel scop (Structural Classification of Proteins). Este indicador hace referencia a las familias proteicasy si alguna familia se repetía era indicativo de que hay zonas conservadas pues hay un origencomún. Hay también otro apartado conocido como EC que está relacionado con la función proteicapero no era muy útil en nuestras investigaciones.
3.9 Programas para generar modelos
Una vez conocemos las secuencias, los templates, los contactos, la estructura secundaria y tenemosuna idea de la terciaria, procedemos a generar los modelos.
3.9.1 Modeller
Modeller fue uno de los programas que más usé en las prácticas. Con él podemos generar modelosde proteínas basándonos en la estructura de uno o más templates. Primero se realiza un alineamientode nuestra secuencia con la del template y luego se incorporan una serie de restricciones al modelopara así poder orientar el resultado hacia donde nosotros deseamos.El programa funciona muy bien con un buen alineamiento, pero se encuentra limitado en las zonasdesalineadas o gaps. Estas son zonas muy variables que Modeller no es capaz de interpretarcorrectamente por lo que debemos evitar aquellos alineamientos con un gran número de gaps o conun gap muy grande ya que en esos casos Modeller genera un loop que puede no parecerse en nada ala estructura del template, lo que no nos interesa en absoluto.
Los alineamientos que introducimos en Modeller están en formato .pirUn ejemplo de este tipo de formato:
32

>P1;T0763 sequence:T0763: : : : :HR9083A, Human, 109 residues: : 0.00: 0.00
>P1; 2mct_A structureX: 2mct_A: 2: A: 102:A:Subdomain of desmoplakin carboxy-terminal domain (DPCT);plakin repeat,, structural protein; 1.80A {Homo sapiens} SCOP d.211.2.1:Homo sapiens:1.80:0.25
En el primer bloque se sitúa nuestro target (T0763) y en el segundo bloque, el template que estamosempleando como molde (2mct_A).
En este caso estamos trabajando con un único template, aunque en ocasiones hemos llegado atrabajar hasta con 3 o 4.Ambas cadenas deben tener el mismo número de elementos sumando residuos y gaps, acabando asía una misma altura (no puede quedar una cadena más larga que la otra).
Una vez teníamos hecho el alineamiento manual, el cual hacía Michael (pues se requiere un altoconocimiento para ajustar los gaps y alinear las distintas secuencias ya que cualquier pequeñocambio podía cambiar la estructura final), procedíamos a utilizar Modeller.
Una vez dentro del programa, tenemos que indicar dónde se encuentra el alineamiento que hemoshecho y dónde se encuentran el template o los templates que hemos utilizado para el mismo.
En alnfile – Introducimos el alineamiento.En knows – el o los templates empleados, en este caso sólo uno (5fd1).En sequence – el nombre que le queremos dar al modelo resultado.
Con sólo estos datos, el programa ya te genera un modelo basándose en el alineamiento creado yusando como moldes los diferentes templates. Se puede generar un modelo que tenga una primeraparte alineada con un template y una segunda alineada con otro creando así un modelo con partes dedos templates, algo que hemos hecho en muchas ocasiones.
Pero la verdadera utilidad de Modeller se encuentra en en las restricciones que podemos añadir:– Restricciones de la estructura secundaria – son verdaderamente útiles y consistían en generar
un alfa-hélice o una lámina beta en una región del modelo. De esta manera forzábamos aciertos residuos a que formaran una estructura secundaria. Lo usábamos principalmentecuando queríamos alargar una alfa-hélice o cuando ciertas regiones desalineadas lesqueríamos dar forma de héice o lámina beta. Bastaba con introducir el aminoácido de inicio
M K K T N K I I F I V F I V I F I G L S Y R H F T N T D K A R M E I S S L S S I D V F K - F N S F S K F S N D KI G V I Y - D E E K L S K F K V I M N S L D T S E G I K K I E V P K D A N I E S F K Y S Y H I Q P N L K Y V E DN N V Y D G Y F L L Y I L V G D S E G K S Y I I F S G T E L S Y V L D K N N T N I L K E I F L N V K K Q Q *
- - - - - - - - - - - - - - - - - - - - - - - - - - - S P I L P K A E N V D S I C I D F T - - - - - - - - - N SI Q K I Y D D S E S I Q K I L S E I A T G K R T E K Q S I Q D Y P S A E - - - - E Y G T I N I E - - - - - - - -- - N N G G M T T M F Y Y E E N - - G K Y Y I E C P - Y K G I Y E I E N N F E D M I - - - - - - - - - - - - *
33

y el del final y elegir entre hélice o lámina beta.
En el primer código estamos generando un alfa hélice entre los residuos 20 y 30, y en los siguientesgeneramos dos lámina beta, uno de los residuos 1 a 6, y el otro de 9 a 14.
Cierto es que no siempre se pueden aplicar estas restricciones pues una región que es porejemplo una alfa-hélice clara, generada a partir de un buen alineamiento, no puedestransformarla en lámina beta porque entre otras cosas estarías desordenando la estructura.
Si quiero añadir una hélice o un strand, lo conveniente es desalinear esa zona, ya que si no, el programa me generará una estructura en esa región tal y como está en el template.
Otra restricción que solíamos emplear era la de juntar varias láminas beta formando así una lámina mayor, algo muy común en las estructuras proteicas tridimensionales. Al igual que antes, escribíamos el aminoácido de la primera lámina beta y el último aminoácido de la última, además debemos añadir si las diferentes láminas se encuentran en sentido paralelo o antiparalelo unas con otras y el número de puentes de hidrógeno que debe haber entre cada par de láminas; y así el programa se encargaba de unirlas todas.
En este ejemplo generamos una sheet con una lámina beta que empieza en el residuo 1 y otra que acaba en el14. Ambas láminas se encuentran en sentido antiparalelo y lo indicamos con el signo negativo que hay antesdel 5, que indica el número de puentes de hidrógeno.
– Puentes disulfuro – como comenté antes, estos puentes puede generar restriccionesimportantes en la estructura, por eso cuando estábamos seguros de su presencia, losincluíamos en Modeller para que el programa los tuviese en cuenta.
Por ejemplo, puente disulfuro entre los residuos 124 y 131:
def special_patches(self, aln): self.patch(residue_type='DISU', residues=(self.residues['124'], self.residues['131']))
– Contactos – Otro de los puntos fuertes del programa es el poder incluir contactos entre losdiferentes aminoácidos. Si sabemos que dos regiones de la estructura deben estar máscercanas, podemos añadir esta información escribiendo un par de aminoácidos, la distanciamedia a la que queremos que se sitúen y la desviación estándar. Esto acercará las regiones alas que pertenezcan dichos aminoácidos moviendo así la estructura a nuestro gusto.
En este caso ponemos en contacto los carbonos alfa del residuo 5 con el del 15, con una distancia media de10 y una desviación estándar de 0.1. Casi siempre hemos trabajado con carbonos alfa pero a veces lo hicimoscon carbonos beta.
34

– Número de modelos – Podemos elegir el número de modelos que queremos como resultado.Nosotros habitualmente trabajábamos con 5 modelos. Como es evidente a mayor número demodelos más tiempo tardaba el programa para procesarlos, por eso cuando queríamos unresultado final empleábamos 5 modelos pero para hacer pequeños cambios, nos bastaba conuno o dos.La diferencia entre los cinco modelos nunca era muy grande pues todos tenían la mismabase (mismo template), tan sólo variaba en las zonas desalineadas.
– Computación – Podíamos pedir un mayor tiempo de computación para cada modelo lo quetambién lleva a un tiempo mayor. Al igual que antes, para pequeños cambios usábamos untiempo de computación menor y para resultados finales trabajábamos con una mayorcomputación. Aun así los cambios tampoco eran muy grandes.
Pero al igual que con el resto de programas, Modeller también tiene algunos problemas a la hora degenerar modelos.
– Nudos – Se pueden generar nudos en los modelos resueltos. Estos nudos sonentrecruzamientos entre cadenas que no son correctos, pues nunca ocurren en la naturaleza.
En esta imagen que pertenece al target T0812 se ve claramente como en la parte superior la cadena naranjase entrelaza con las azules, a eso se le conoce como nudo y debe evitarse.
Para deshacer un nudo podemos cambiar el alineamiento o de manera más sencilla, podemosmover la estructura con la restricción de contactos sin alterarla demasiado o a veces es suficiente con alargar o acortar una hélice o lámina beta si esta se ve implicada en el nudo.
– Clashes – También puede ocurrir que al modificar la estructura se produzcan colisiones otambién llamados clashes (superposición de aminoácidos) entre diferentes regiones de laestructura. Para solucionarlo basta con hacer lo mismo que con los nudos.
35

En este caso vemos lo que podríamos denominar un completo ovillo de lana con cadenas que se entrecruzansin orden alguno. Pertenece al modelo generado por el servidor MUFOLD del target T0826.
– Huecos en el modelo – Hay veces en los que pueden aparecer huecos o ausencias dedeterminadas regiones en el modelo final. Esto puede deberse a que el template que hemosutilizado de la base de datos de PDB ya venía con este hueco debido a que son regiones queno han cristalizado. Para solventar dicho problema basta con añadir gaps en el alineamiento que correspondancon estos residuos que faltan de esta manera evitaremos la presencia de huecos en nuestromodelo final.Si los huecos aparecen en el modelo de Modeller, pero no estaban presentes en el template,es debido a un error del programa.
3.9.2 FoldIt
FoldIt es una especie de juego que puntúa la apariencia de tu modelo. A mayor número de clashesentre residuos y entre cadenas laterales de residuos, menor puntuación, por lo que hay que irmoviendo la estructura hasta alcanzar una puntuación aceptable.Este programa está basado en la plataforma Rosetta para realizar los cálculos, por lo que funcionabastante bien.
Esta herramienta la usé menos que Modeller pero en ciertas ocasiones recurrimos a ella, sobre todoa la hora de hacer grandes cambios en la molécula (en una ocasión teníamos una hélice situada porencima del modelo y queríamos situarla justo por debajo de la estructura) y cuando deseábamosreducir el número de clashes de nuestro modelo.Solíamos usar FoldIt como complemento de Modeller.
36

En este ejemplo vemos una proteína muy simple donde las bolas rojas representan las colisiones que se estánproduciendo. A mayor tamaño de la bola, mayor es la colisión.
3.9.3 Swiss-Model
Swiss-Model es un servidor dedicado al modelado de estructuras tridimensionales en proteínas. Laselección del template, el alineamiento y la construcción del modelo lo hace automáticamente elservidor [23]. La secuencia proteica y las bases de datos para la construcción de la estructura sonaccesibles desde el workspace [24].
En alguna escasa ocasión (en uno o dos targets) hemos hecho uso de los modelos que proponíaSwiss-Model antes que los obtenidos con Modeller. Esto fue debido a que Modeller intentaminimizar los errores en todo el modelo lo que generalmente es bueno porque hace uso deestadísticas físicas. Pero esto puede suponer un cambio en el backbone o esqueleto de la estructuraque no nos interesaba.Swiss_Model no produce cambio alguno en el backbone así que si el alguna ocasión quisimoscambiar algo de un modelo sin alterar el esqueleto hicimos uso entonces de Swiss-Model, aunquecomo ya dije lo usamos en muy pocas ocasiones.
3.9.4 EV fold
Es un programa que genera modelos basándose en contactos y es útil cuando hay hélicestransmembrana. Pero los resultados obtenidos con este programa no suelen ser muy buenos ytenemos que mejorarlos con modeller.
3.10 Visualización de modelos
PYMOL
Programa estrella a la hora de visualizar los diferentes modelos. Junto con Modeller fue el programaque más llegué a usar. Es realmente útil debido a la cantidad de posibilidades que tiene. Secomplementa muy bien con Modeller pues una vez generados los diferentes modelos pasábamos avisualizarlos en Pymol, y era en este programa donde podíamos encontrar nudos, clashes o huecos.
Una vez en Pymol podíamos visualizar las proteínas en modo “Cartoon” que era el que más
37

usábamos pues dibuja las alfa-hélices en forma de hélices y las lámina betas en forma de flechasque indican la dirección de la secuencia, de esta manera dos flechas con sentidos opuestos indicabados láminas beta antiparalelas. La visión global de la proteína era mucho más intuitiva de estamanera. Todos los ejemplos de targets que he puesto hasta ahora están sacados de pymol en versiónCartoon.Aunque a veces cambiábamos la visión a “sticks” de manera que se pueden apreciar mucho másclaramente los clashes, o a “spheres” donde se visualiza la proteína en su conjunto con los espaciosque deja lo que puede resultar interesante a la hora de conocer espacios donde podamos situar lasdiferentes cadenas o para conocer dónde pueden unirse diversos ligandos.
A la izquierda está la visualización en sticks y a la derecha en esferas. Los colores representan los diferentestipos de átomos que hay en cada residuo. El verde son Carbonos, el azul son Nitrógenos, el rojo sonOxígenos y el anaranjado Azufres.
Pero podemos cambiar estos colores a nuestro antojo, y no sólo el de los átomos si no también en elde los residuos, lo cual nos puede resultar realmente útil ya que por ejemplo lo usábamos paracolorear residuos hidrofóbicos, los cuales debían aparecer en la zona interna de la estructura; o paracolorear las regiones que aparecían como gaps en el template, para ver a qué zonas de nuestromodelo correspondían.Además en Pymol se puede ver la secuencia de aminoácidos por lo que si coloreas una región secolorea a su vez la secuencia y así puedes ver de manera sencilla a qué aminoácido corresponde esaregión.
38
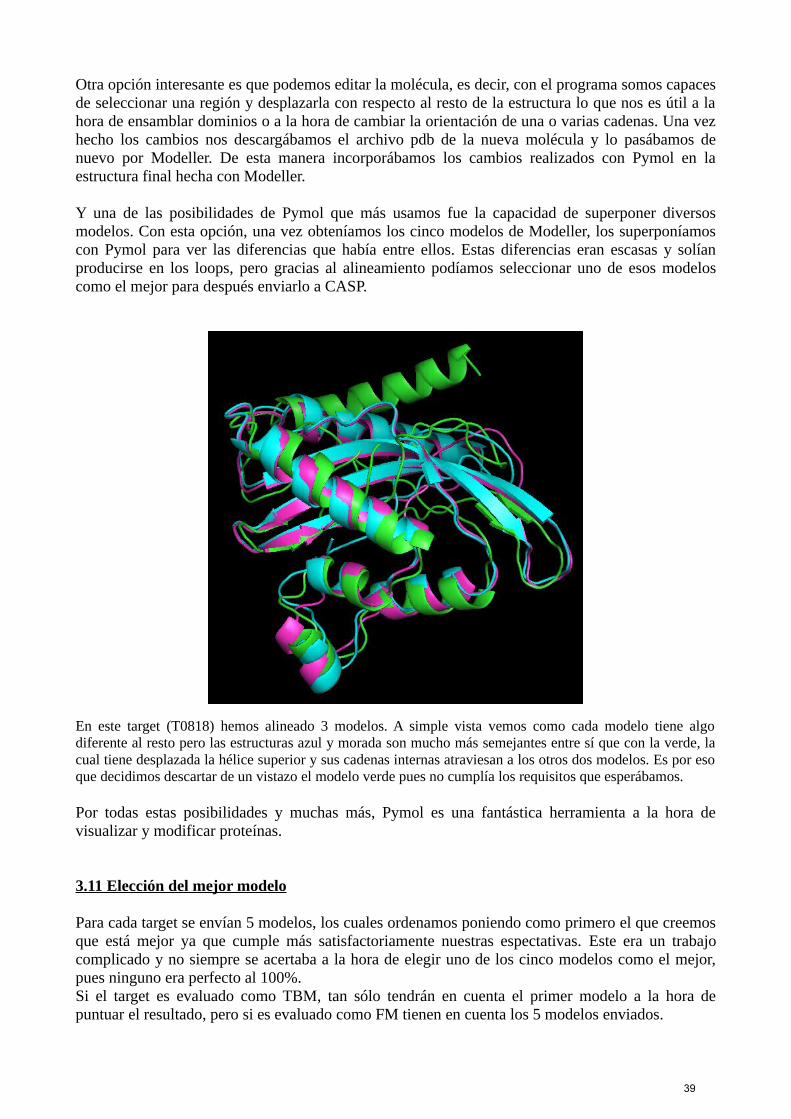
Otra opción interesante es que podemos editar la molécula, es decir, con el programa somos capacesde seleccionar una región y desplazarla con respecto al resto de la estructura lo que nos es útil a lahora de ensamblar dominios o a la hora de cambiar la orientación de una o varias cadenas. Una vezhecho los cambios nos descargábamos el archivo pdb de la nueva molécula y lo pasábamos denuevo por Modeller. De esta manera incorporábamos los cambios realizados con Pymol en laestructura final hecha con Modeller.
Y una de las posibilidades de Pymol que más usamos fue la capacidad de superponer diversosmodelos. Con esta opción, una vez obteníamos los cinco modelos de Modeller, los superponíamoscon Pymol para ver las diferencias que había entre ellos. Estas diferencias eran escasas y solíanproducirse en los loops, pero gracias al alineamiento podíamos seleccionar uno de esos modeloscomo el mejor para después enviarlo a CASP.
En este target (T0818) hemos alineado 3 modelos. A simple vista vemos como cada modelo tiene algodiferente al resto pero las estructuras azul y morada son mucho más semejantes entre sí que con la verde, lacual tiene desplazada la hélice superior y sus cadenas internas atraviesan a los otros dos modelos. Es por esoque decidimos descartar de un vistazo el modelo verde pues no cumplía los requisitos que esperábamos.
Por todas estas posibilidades y muchas más, Pymol es una fantástica herramienta a la hora devisualizar y modificar proteínas.
3.11 Elección del mejor modelo
Para cada target se envían 5 modelos, los cuales ordenamos poniendo como primero el que creemosque está mejor ya que cumple más satisfactoriamente nuestras espectativas. Este era un trabajocomplicado y no siempre se acertaba a la hora de elegir uno de los cinco modelos como el mejor,pues ninguno era perfecto al 100%.Si el target es evaluado como TBM, tan sólo tendrán en cuenta el primer modelo a la hora depuntuar el resultado, pero si es evaluado como FM tienen en cuenta los 5 modelos enviados.
39

Para facilitar la selección del mejor modelo, generábamos rankings basados en los contactos porconsenso y rankings basados en los contactos por coevolución, con los modelos creados por losdiversos servidores más los que nosotros construíamos. De todos nuestros modelos nos quedábamoscon 5, entre los cuales elegíamos uno como el mejor, pero podíamos llegar a generar entre 30 y 40modelos fácilmente para determinados targets.Estos rankings ordenaban los modelos de mejor a peor según las distancias entre los carbonos alfapara cada contacto, es decir, el modelo situado en primera posición era el que más se ajustaba a loscontactos predichos pues la suma de la distancia entre los mismos era la menor. Los rankings basados en los contactos por consenso siempre podían llevarse a cabo pues siempre sedisponía de los modelos generados por los servidores pero con los rankings basados en contactospor coevolución, si no disponíamos de suficientes secuencias o lo suficientemente variadas, nopodíamos generarlos, pues carecíamos de este tipo de contactos.
Una vez teníamos los rankings, los comparábamos para seleccionar nuestros mejores modelos, elproblema venía en que los modelos que quedaban en buena posición en un ranking, no lo hacían enel otro y viceversa, por lo que teníamos que manejar toda la información de la que disponíamos pararealizar una buena selección. Inicialmente elegíamos los que a primera vista en los rankings,parecían nuestros mejores modelos y los descargábamos en pymol junto con los mejores modelosde los servidores y luego los superponíamos para estudiar las semejanzas. La superposición con otros modelos de servidores resultaba muy útil pues si en ocasiones teníamosdudas de cómo colocar una determinada región de nuestro modelo, podíamos ver qué habían hecholos servidores con mejor puntuación. Al compararlos nos fijábamos en si los residuos hidrofóbicosquedaban en la parte interior de los modelos, en si había nudos o clashes, en las longitudes de lasalfa hélice y láminas beta, en las zonas conservadas en los alineamientos … y si nuestros modelosno nos convencían, los modelábamos de nuevo con modeller y volvíamos a introducirlos en losrankings, hasta conseguir un modelo que nos convenciese. De esta forma mejorábamos nuestrosmodelos e íbamos ascendiendo posiciones en el ranking hasta obtener un puesto lo suficientementebueno como para seleccionar ese modelo como número 1.
A la hora de enviar los cinco modelos seleccionábamos:Como primer modelo el humano, es decir, aquel modelo que pensábamos nosotros que era el mejorbasándonos en la información de los rankings y las superposiciones.Como segundo modelo nuestro mejor modelo en el ranking de Model Contacts.Como tercer modelo el de Psicov si había pocas secuencias o el de Plmdca si había muchas.Como cuarto modelo el de Simone.Y por último, el modelo de Pcons.
40

4. RESULTADOS
En esta edición de CASP se han registrado un total de 207 grupos diferentes, 123 de los cuálespertenecen a grupos humanos y los 84 restantes son servidores. Este año el número de gruposhumanos ha ascendido con respecto a ediciones pasadas, mientras que el número de servidores hadescendido.Los servidores que copan los altos puestos son siempre los mismos y puesto que mantener unservidor no es para nada sencillo, aquellos servidores con puntuaciones bajas tienden a desaparecer.Los mejores son Zhang-Server y QUARK, a gran distancia de un segundo bloque compuesto pormyprotein, BAKER y MULTICOM, y un tercer bloque algo más alejado con RaptorX y el servidorde Hhpred. El resto de servidores como FFAS, FLOUDAS y SAM-T08 entre otros se encuentranmuy alejados de los iniciales.
El número de targets publicados fue de un total de 100, de los cuales tuvimos que trabajar con 55,que fueron los incluidos para todos los grupos. El resto fueron resueltos únicamente por losservidores. Estos números son semejantes a los de ediciones anteriores.Además se analizan los dominios por separado y aunque hayamos trabajado con 55 targets, enrealidad se están analizando 78 dominios en total.
Los resultados finales no estuvieron completos hasta principios de diciembre por lo que hastaentonces era imposible conocer todos los modelos definitivos, los GDT-plots y los resultados en losdiferentes rankings. Aun así, cuando empezaron a salir los primeros modelos resueltos en el mes deseptiembre, Michael y Juan elaboraron diferentes rankings donde calculaban la puntuación obteniday la comparaban con la del resto de grupos servidores, pues aún no conocíamos los modelos de losgrupos humanos. No era este un resultado definitivo pero nos servía como idea inicial de cómohabíamos puntuado ese modelo con respecto al resto.
Esta imagen pertenece a un ranking que elaboramos para el target T0761.
41
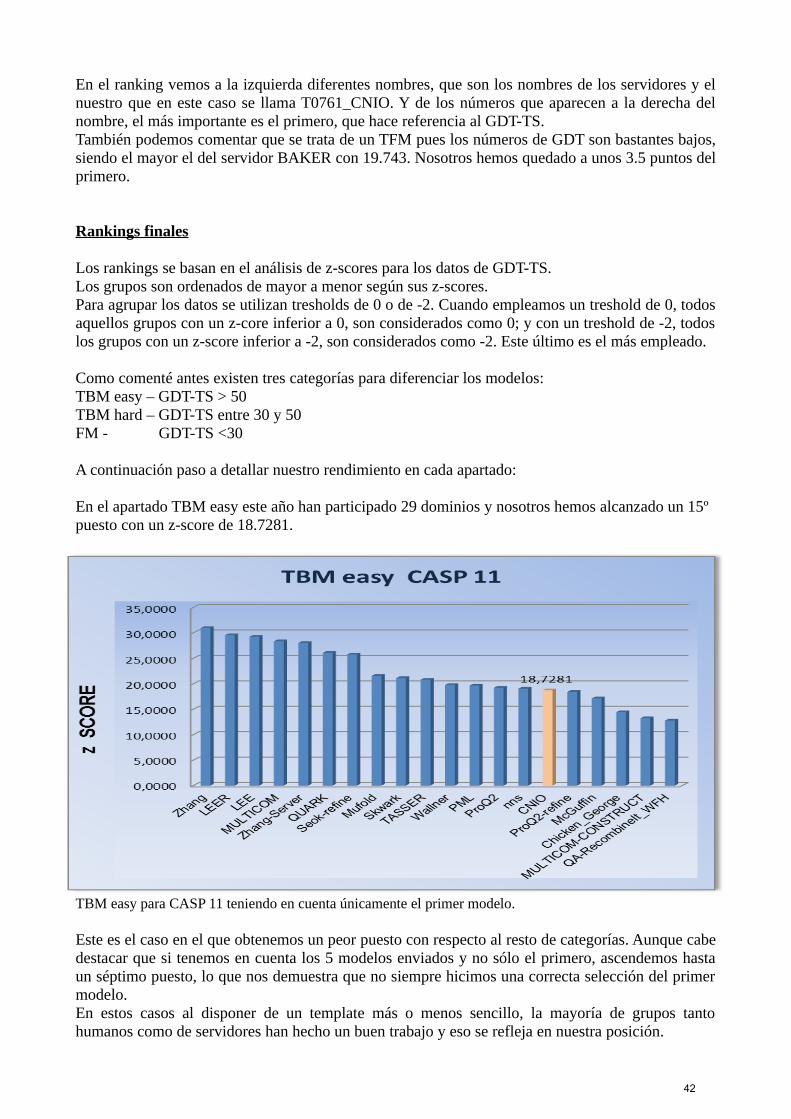
En el ranking vemos a la izquierda diferentes nombres, que son los nombres de los servidores y elnuestro que en este caso se llama T0761_CNIO. Y de los números que aparecen a la derecha delnombre, el más importante es el primero, que hace referencia al GDT-TS.También podemos comentar que se trata de un TFM pues los números de GDT son bastantes bajos,siendo el mayor el del servidor BAKER con 19.743. Nosotros hemos quedado a unos 3.5 puntos delprimero.
Rankings finales
Los rankings se basan en el análisis de z-scores para los datos de GDT-TS. Los grupos son ordenados de mayor a menor según sus z-scores.Para agrupar los datos se utilizan tresholds de 0 o de -2. Cuando empleamos un treshold de 0, todosaquellos grupos con un z-core inferior a 0, son considerados como 0; y con un treshold de -2, todoslos grupos con un z-score inferior a -2, son considerados como -2. Este último es el más empleado.
Como comenté antes existen tres categorías para diferenciar los modelos: TBM easy – GDT-TS > 50TBM hard – GDT-TS entre 30 y 50FM - GDT-TS <30
A continuación paso a detallar nuestro rendimiento en cada apartado:
En el apartado TBM easy este año han participado 29 dominios y nosotros hemos alcanzado un 15º puesto con un z-score de 18.7281.
TBM easy para CASP 11 teniendo en cuenta únicamente el primer modelo.
Este es el caso en el que obtenemos un peor puesto con respecto al resto de categorías. Aunque cabedestacar que si tenemos en cuenta los 5 modelos enviados y no sólo el primero, ascendemos hastaun séptimo puesto, lo que nos demuestra que no siempre hicimos una correcta selección del primermodelo.En estos casos al disponer de un template más o menos sencillo, la mayoría de grupos tantohumanos como de servidores han hecho un buen trabajo y eso se refleja en nuestra posición.
42
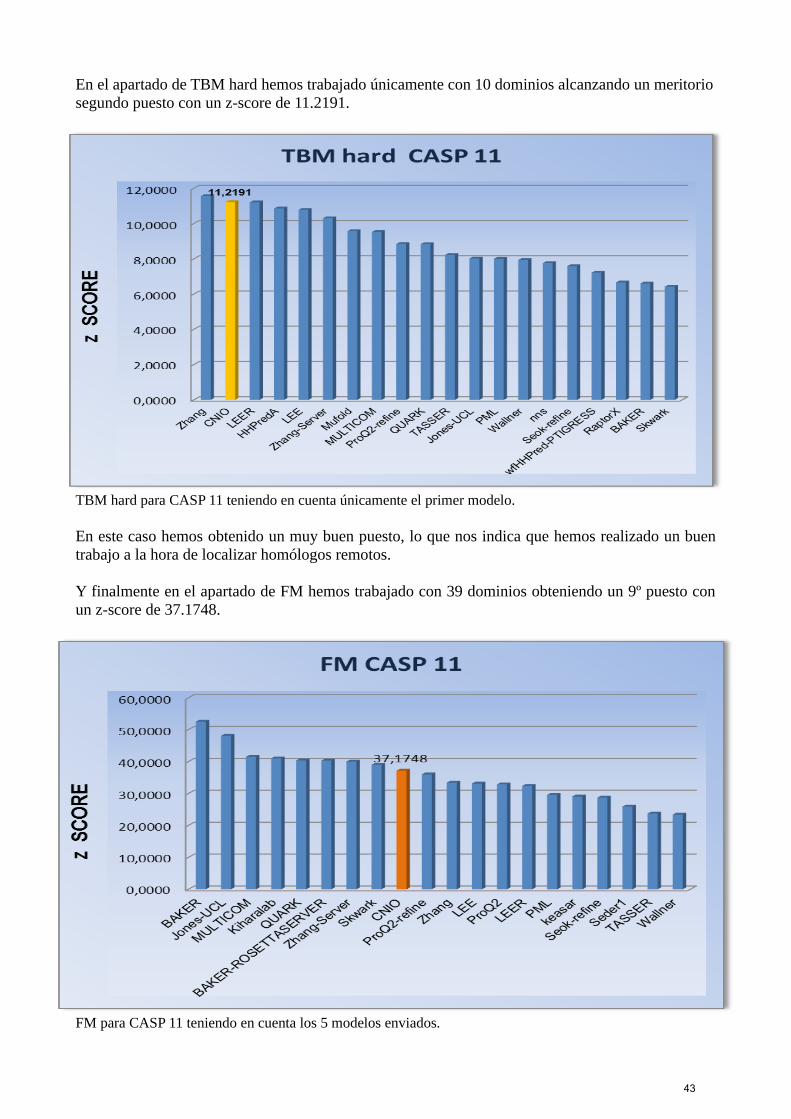
En el apartado de TBM hard hemos trabajado únicamente con 10 dominios alcanzando un meritoriosegundo puesto con un z-score de 11.2191.
TBM hard para CASP 11 teniendo en cuenta únicamente el primer modelo.
En este caso hemos obtenido un muy buen puesto, lo que nos indica que hemos realizado un buentrabajo a la hora de localizar homólogos remotos.
Y finalmente en el apartado de FM hemos trabajado con 39 dominios obteniendo un 9º puesto conun z-score de 37.1748.
FM para CASP 11 teniendo en cuenta los 5 modelos enviados.
43

Basándonos en estos resultados, podemos afirmar que hemos realizado un buen trabajo, puesnuestras posiciones con respecto al resto de grupos son elevadas.En la categoría TBM easy nos encontramos por detrás de grupos como Zhang (líder de laclasificación), MULTICOM, QUARK (servidor del grupo de Zhang), Zhang-Server o Mufold entreotros. Uno de los motivos por los Zhang-Server obtiene buena puntuación es debido a que es capaz decompactar mucho sus modelos y es por ello que los teníamos en cuenta a la hora de generar losnuestros.En los TBM hard tan sólo estamos por detrás del grupo de Zhang.Y en los FM sale victorioso el grupo de BAKER y con un sexto puesto para su servidor BAKER-ROSETTASERVER, lo cual era de esperar pues trabaja muy bien en el modelado proteico ab initioy de novo.
Comparación con CASP 10
Gráfica que compara las categorías TBM easy, TBM hard y FM en tres bloques diferentes (model 1: posiciónobtenida teniendo en cuenta sólo el primer modelo, best scores: posición obtenida teniendo en cuenta los mejores modelos y domains: el número de dominios) tanto en CASP 10 como en CASP 11.
Centrándonos en el apartado “model 1” no se puede decir a simple vista si esta edición hemosobtenido mejores resultados que en la edición pasada. Pues aunque en la resolución de dominiosTBM easy alcanzamos un peor puesto, mejoramos con respecto a TBM hard, aunque en ninguno delos dos casos la diferencia sea muy abultada.Los FM los tenemos que evaluar en el apartado “best scores”, pues en esta categoría se tienen encuenta los 5 modelos y con respecto a CASP 10, mejoramos en 6 posiciones (del 15º al 9º).La principal diferencia con respecto a la edición pasada es el tipo de dominios con los que hemostrabajado. Si nos fijamos ahora en el bloque “Domains”, aunque los TBM hard son más o menos elmismo número en ambos CASP (10 y 13), la diferencia con respecto a los otros dos grupos esnotable. En CASP 10 hubo 43 dominios TBM easy y tan sólo 14 FM, mientras que este año seinvierten los datos, habiendo 29 TBM easy y 39 FM, lo que en este caso es casi el triple dedominios FM que en la edición anterior.
TBM EASY
TBM HARD
FM TBM EASY
TBM HARD
FM TBM EASY
TBM HARD
FM
MODEL I BEST SCORES DOMAINS
0
5
10
15
20
25
30
35
40
45
15
2
19
75
9
29
10
39
8 7
22
4 4
15
43
13 14
COMPARATIVA CASP 10 / 11
CASP 11
CASP 10
44
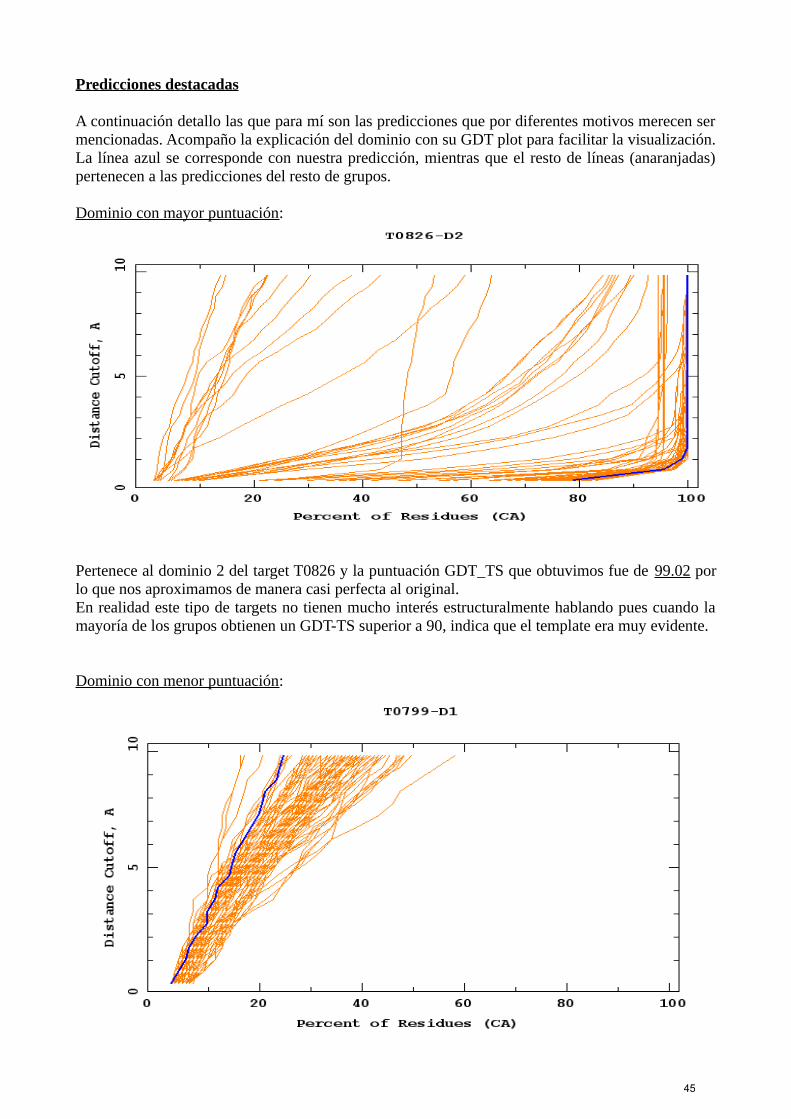
Predicciones destacadas
A continuación detallo las que para mí son las predicciones que por diferentes motivos merecen sermencionadas. Acompaño la explicación del dominio con su GDT plot para facilitar la visualización.La línea azul se corresponde con nuestra predicción, mientras que el resto de líneas (anaranjadas)pertenecen a las predicciones del resto de grupos.
Dominio con mayor puntuación:
Pertenece al dominio 2 del target T0826 y la puntuación GDT_TS que obtuvimos fue de 99.02 porlo que nos aproximamos de manera casi perfecta al original.En realidad este tipo de targets no tienen mucho interés estructuralmente hablando pues cuando lamayoría de los grupos obtienen un GDT-TS superior a 90, indica que el template era muy evidente.
Dominio con menor puntuación:
45
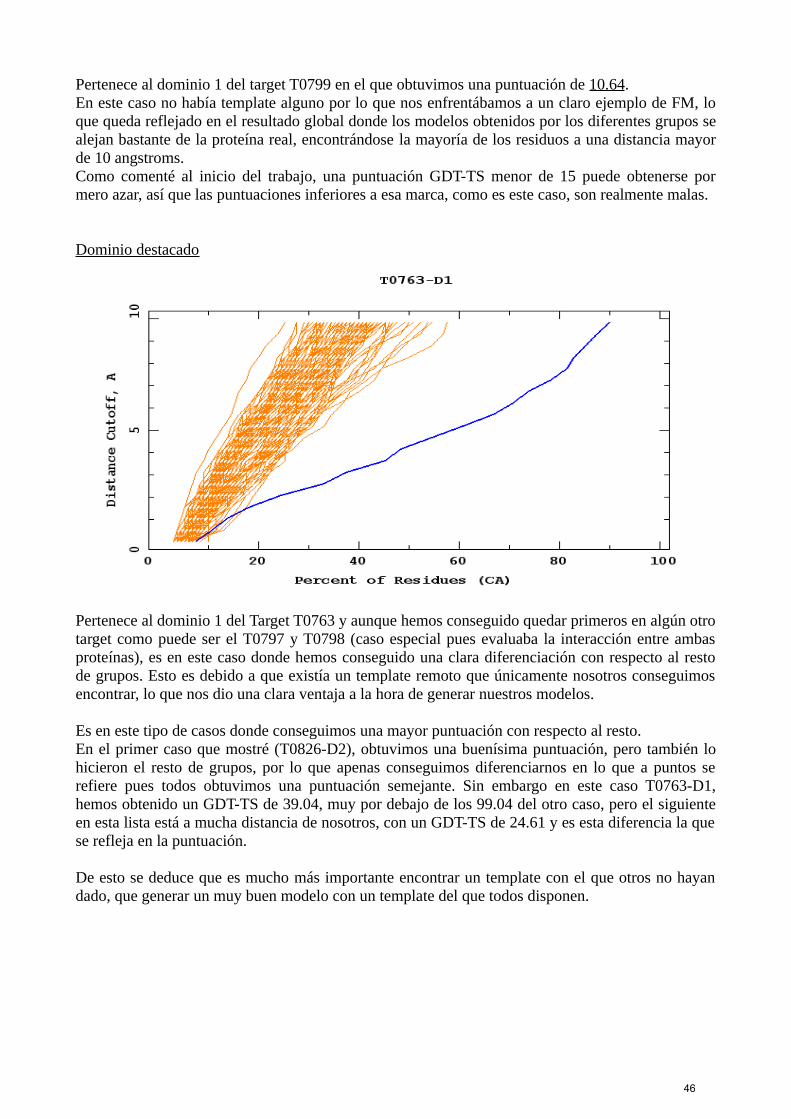
Pertenece al dominio 1 del target T0799 en el que obtuvimos una puntuación de 10.64.En este caso no había template alguno por lo que nos enfrentábamos a un claro ejemplo de FM, loque queda reflejado en el resultado global donde los modelos obtenidos por los diferentes grupos sealejan bastante de la proteína real, encontrándose la mayoría de los residuos a una distancia mayorde 10 angstroms.Como comenté al inicio del trabajo, una puntuación GDT-TS menor de 15 puede obtenerse pormero azar, así que las puntuaciones inferiores a esa marca, como es este caso, son realmente malas.
Dominio destacado
Pertenece al dominio 1 del Target T0763 y aunque hemos conseguido quedar primeros en algún otrotarget como puede ser el T0797 y T0798 (caso especial pues evaluaba la interacción entre ambasproteínas), es en este caso donde hemos conseguido una clara diferenciación con respecto al restode grupos. Esto es debido a que existía un template remoto que únicamente nosotros conseguimosencontrar, lo que nos dio una clara ventaja a la hora de generar nuestros modelos.
Es en este tipo de casos donde conseguimos una mayor puntuación con respecto al resto. En el primer caso que mostré (T0826-D2), obtuvimos una buenísima puntuación, pero también lohicieron el resto de grupos, por lo que apenas conseguimos diferenciarnos en lo que a puntos serefiere pues todos obtuvimos una puntuación semejante. Sin embargo en este caso T0763-D1,hemos obtenido un GDT-TS de 39.04, muy por debajo de los 99.04 del otro caso, pero el siguienteen esta lista está a mucha distancia de nosotros, con un GDT-TS de 24.61 y es esta diferencia la quese refleja en la puntuación.
De esto se deduce que es mucho más importante encontrar un template con el que otros no hayandado, que generar un muy buen modelo con un template del que todos disponen.
46

5. Discusión
A raíz de los resultados obtenidos podemos afirmar que nuestros métodos de búsqueda y sobre todoaquellos basados en la predicción de contactos tanto por coevolución como por consenso, resultanefectivos.
Cada vez más se van incorporando métodos de predicción de contactos a la hora de construir yevaluar los diferentes modelos de estructuras proteicas.Por ejemplo el grupo Jones usó el predictor de contactos CONSIP 2 que se basa en un método deredes neuronales las cuales incorporan modelos de tres predictores diferentes basados en lacoevolución, y ha dado muy buenos resultados este año.
Y ya que CASP tiene como principal objetivo evaluar los diferentes métodos de predicción deestructuras, me gustaría hablar del perfecto trabajo que realizan buscadores como HHBlits oHHMER, muy por encima de BLAST o PsiBLAST, los cuales tienen un método de búsqueda máspobre y en experimentos como estos resultan poco útiles.
A la hora de alinear me sigo quedando con HHBlits y HHMER.
Si buscamos el mejor programa para buscar templates, ese es Hhpred, pues a parte de tener un buenmétodo de búsqueda, realiza sus comparaciones frente a unas buenas bases de datos.
Si tengo que escoger un programa que elabore modelos estructurales, me quedo sin duda conModeller, el cual está muy por encima de sus contrincantes, aunque es cierto que a la hora derealizar grandes cambios estructurales, se trabajaba más fácilmente con FoldIt, pues Modellerdependía mucho del alineamiento introducido.
Para visualizar estructuras no hay ningún programa mejor que Pymol, por su versatilidad, sufacilidad a la hora de manejarlo, sus innumerables funciones y su detalle a la hora de mostrar laimagen.
Y por último quiero destacar la complicada labor de selección del mejor modelo basándonos enrankings construidos a partir de los contactos predichos y el resto de estructuras de servidores.Cierto es que en este aspecto se podría mejorar, pues no siempre se elije el mejor modelo posible.
Sinceramente creo que este tipo de experimentos son realmente beneficiosos a la hora de evaluar losdistintos métodos posibles y de mejorarlos para un futuro.
47

6. Conclusión
Nuestro trabajo como participantes en CASP 11 puede resultar en apariencia repetitivo puesseguíamos el mismo procedimiento para cada target, pero en cada uno de ellos encontrábamos algodiferente que hacía que actuásemos de forma distinta para obtener los mejores modelos.
Empezamos siempre buscando secuencias homólogas a la nuestra con BLAST, PsiBLAST,HHMER, HHblits e incluso Pfam. Obtenemos los alineamientos con HHMER y HHblits. Estudiamos las zonas conservadas yreunimos información de las familias y la historia evolutiva con diferentes programas y papers.También buscamos los posibles templates en Hhpred.Según el número de secuencias que encontremos y de la variabilidad de las mismas podemosaplicar los métodos de predicción de contactos por coevolución. Y para los contactos por consenso,nos basamos en los modelos de los servidores.Una vez que tenemos alineamientos, templates e información de los contactos, los empleamos conModeller para generar nuestros modelos, los cuales evaluamos con los rankings construidos basadosen los contactos.Visualizamos con Pymol donde realizaremos pequeños cambios o repetiremos el proceso.
Esto sería un resumen si el target es un TBM easy. Pero si es TBM hard, debemos ampliar nuestrabúsqueda con la secuencia a la que pertenece nuestro target, si es que la hay, investigar sobre lafamilia o superfamilia a la que pertenece, buscar templates con alineamientos o perfiles en lugar decon nuestro target, en definitiva realizar una búsqueda más exhaustiva.
Y por último si trabajamos con un FM, tan sólo podemos basarnos en los modelos de los servidores,en la información adquirida y en nuestra intuición para modelar ciertas regiones.
Este era nuestro día a día a la hora de generar modelos para nuestros targets.
Por otra parte, en la “lucha” mantenida entre grupos humanos y servidores, salen victoriosos losgrupos humanos, pero no con una diferencia clara. Hay muchos servidores con muy buenosresultados como Zhang-Server en los TBM y ROSETTA-SERVER para los FM.Los grupos humanos tienen un ventaja clara a la hora de interpretar los datos y deducir cuales sonlos mejores para cada caso.En nuestro caso, Michael realizaba a mano los alineamientos entre targets y templates de unamanera más efectiva que el trabajo realizado por los servidores. Pero esto no quita que en un futurono muy lejano, los servidores mejoren de tal manera que acabarán siendo muy superiores a losgrupos humanos. Pues aunque ya lo son en multitud de aspectos como a la hora de alinear grancantidad de secuencias, buscar en bases de datos o manejar simplemente grandes cantidades deinformación, les falta ese “toque” humano de interpretación de los datos.
Para concluir me gustaría decir que hay muchos métodos y programas que han mejorado conrespecto a años pasados y lo seguirán haciendo, pues la predicción de estructuras proteicas meparece una interesante labor científica con multitud de posibilidades, diferentes utilidades y con unimportante futuro.
48

7. BIBLIOGRAFÍA
[1] Adam Zemla. “LGA: a method for finding 3D similarities in protein structures.” Nucleic Acids Research, Jul 1, 2003; 31(13): 3370–3374.
[2] Yang Zhang . “Protein structure prediction: when is it useful? ”Current Opinion in Structural Biology, 2009
[3] Chothia C, Lesk AM. “The relation between the divergence of sequence and structure inproteins.”Embo J. 1986; 5(4):823–826.
[4] Mathews, Van Holde, Ahern. Bioquímica 2002; 200 [5] von Heijne G. “Signal sequences: The limits of variation”. J Mol Biol, Jul de 1985; 184 (1): 99–105
[6] Liu, J; Zheng Q; Deng Y; Cheng CS; Kallenbach NR; Lu M. “A seven-helix coiled coil”PNAS 2006; 103 (42): 15457–15462
[7] Kobe B, Deisenhofer J . “The leucine-rich repeat: a versatile binding motif”. Trends Biochem. Sci. (October 1994); 19 (10): pp. 415–21
[8] Enkhbayar P, Kamiya M, Osaki M, Matsumoto T, Matsushima N . “Structural principles ofleucine-rich repeat (LRR) proteins”. Proteins (February 2004); 54 (3): pp. 394–403.
[9] Wolanin PW, Thomason PA, Stock JB. “Histidine protein kinases: key signal transducers outsidethe animal kingdom”. Genome Biology (200); 3 (10): reviews3013.1–3013.8.
[10] Lima Mde, F; Eloy, NB; Pegoraro, C; Sagit, R; Rojas, C; Bretz, T; Vargas, L; Elofsson, A; deOliveira, AC; Hemerly, AS; Ferreira, PC. “Genomic evolution and complexity of the Anaphase-promoting Complex (APC) in land plants.”. BMC plant biology (Nov 18, 2010); 10: 254
[11] Lengauer T. “Computational methods for biomolecular docking”. Current Opinion in Structural Biology. 1996 6;6(3):402-406.
[12] Sonnhammer EL, Eddy SR, Durbin R.“Pfam: a comprehensive database of protein domainfamilies based on seed alignments”.Proteins. 1997 Jul;28(3):405-20.
[13] M. A. Marti-Renom, Stuart AC, Fiser A, Sánchez R, Melo F, Sali A. “Comparative proteinstructure modeling of genes and genomes”.Annu. Rev. Biophys. Biomol. Struct. (2000); 29: 291-325
[14] Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. “GappedBLAST and PSI-BLAST: a new generation of protein database search programs”.Nucleic Acids Res. 1997 Sep 1;25(17):3389-402.
49

[15] D. J. Ayers, P. R. Gooley, A. Widmer-Cooper, and A. E. Torda. “Enhanced protein foldrecognition using secondary structure information from NMR”.Protein Sci. May 1999; 8(5): 1127–1133.
[16] Baker D, Sali A. “Protein structure prediction and structural genomics”.Science. 2001 Oct 5; 294(5540):93-6.
[17] Michael Remmert, Andreas Biegert, Andreas Hauser, Johannes Söding. “HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment”Nature Methods 9, 173–175
[18] Kabsch W, Sander C.“Dictionary of protein secondary structure: pattern recognition ofhydrogen-bonded and geometrical features”.Biopolymers. 1983 Dec; 22(12):2577-637.
[19] Michele Clamp, James Cuff, Stephen M. Searle and Geoffrey J. Barton. “The Jalview Javaalignment editor”Bioinformatics 2003; Pp 426-427
[20] Michael L. Tress and Alfonso Valencia. “Predicted residue–residue contacts can help thescoring of 3D models”Proteins , 2010
[21] Venkatarajan Mathura; Pandjassarame Kangueane. “Bioinformatics: A Concept-BasedIntroduction”. Springer (11 October 2008); pp. 66–67.
[22] - Kurowski MA1, Bujnicki JM. “GeneSilico protein structure prediction meta-server”.Nucleic Acids Res. 2003 Jul 1;31(13):3305-7.
[23] - Schwede T, Kopp J, Guex N, Peitsch MC. “SWISS-MODEL: an automated protein homology-modeling server”. Nucleic Acids Research 2003; 31 (13): 3381–3385
[24] - Arnold K, Bordoli L, Kopp J, and Schwede T. “The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling”. Bioinformatics 2009; 22 (2): 195–201.
50