anemia hemolitik -2014
DESCRIPTION
anemiaTRANSCRIPT

ANEMIA HEMOLITIK
Nining Sri WuryaningsihFak. Kedokteran UKDW

Anemia Hemolitik
Anemia karena peningkatan destruksi eritrositTerjadi hiperplasi eritropoetik & perluasan anatomis tulangSS tl : 6 – 8 xNRetikulosit meninggi
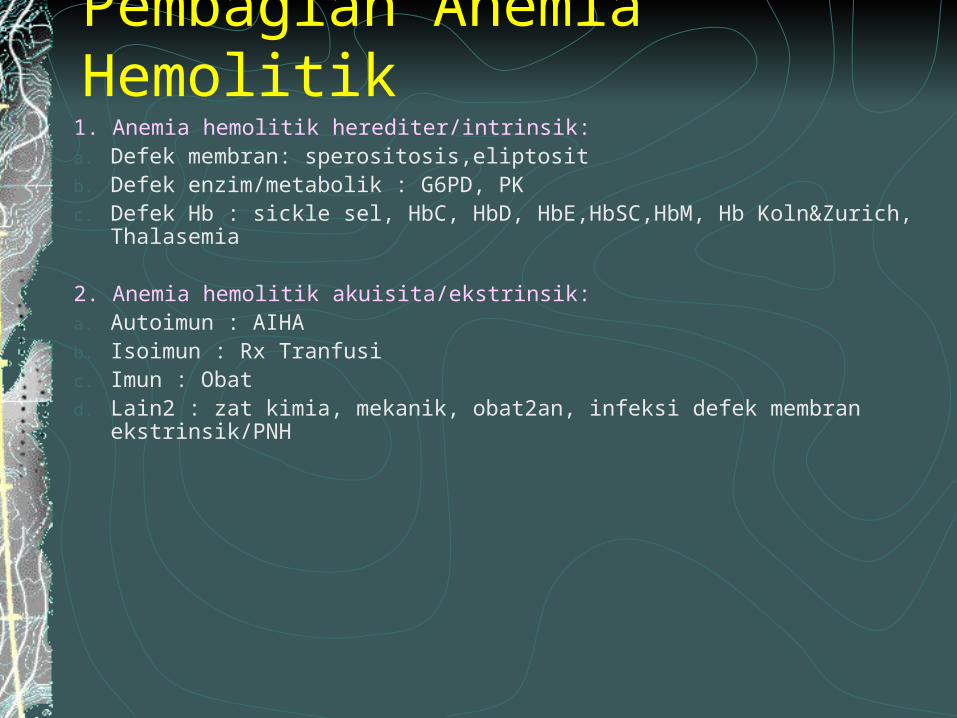
Pembagian Anemia Hemolitik1. Anemia hemolitik herediter/intrinsik:a. Defek membran: sperositosis,eliptositb. Defek enzim/metabolik : G6PD, PKc. Defek Hb : sickle sel, HbC, HbD, HbE,HbSC,HbM, Hb Koln&Zurich, Thalasemia
2. Anemia hemolitik akuisita/ekstrinsik:a. Autoimun : AIHAb. Isoimun : Rx Tranfusic. Imun : Obatd. Lain2 : zat kimia, mekanik, obat2an, infeksi defek membran ekstrinsik/PNH

Gambaran dasarDestruksi eritrosit abnormal / dipercepatPemecahan Hb meningkat kadar bilirubin ,urobilinogen urin dan faeces Sumsum tulang ; hiperplasi eritroid dgn produksi eritrosit dipercepat retikulositosis & makrositosis ringan Berat ringannya anemia ditentukan keseimbangan antara destruksi eritrosit dan kompensasi su-tul

Sferositosis herediterAutosomal dominanCacad struktur membran, protein spektrin utk bentuk bikonkaf, Sel menjadi sferis, mati lebih diniDarah tepi: eritrosit tampak sferis dan beberapa diantaranya kecil “mikrosferosit”Anemia ringan pada kasus kompensasi; MCV biasanya normal. retikulositosisDirek coomb test negatifAutohemolitik akibat fragilitas osmotik yg meningkatCr 51 destruksi limpa t>>

Gambaran klinikKebanyakan penderita sebelum usia pubertasKadang ditemukan secara kebetulan, diagnose lambat ditegakkanSplenomegaliAnemia dan ikterus berat ringannya tergantung kecepatan hemolisis dan derajat kompensasi

Defek enzim G6 PD• Diturunkan “Sex linked” resesil lewat gene pada kromosom X (seperti hemofili)
•Faktor pencetus : infeksi, peny akut, obat2, kacang fava
•30% menyerang orang negro ( yang berasal dari Mediteranea)
•Penyakit timbul pada pria homozygote dan wanita heterizygote

PATOGENISISAktivitas G6PD eritrosit menurun dengan cepat, juga usia eritrosit menurunMetabolisme glukose menurunRasio NADPH:NADP dan GSH: GSSG berkurangTerjadi oksidasi Hb dan sulhidril dalammembranIntegritas eritrosit terganggu

HEMATOLOGI
Anemia normokromnormositerSelama episode hemolitik retikulositosis, eritrosit berintiTampak adanya Heinz bodiesAnemia bisa berat pada hemolisis akut yang tidak terkompesir

Defek enzim PKResesif autosomal homozigot Hemokisis bermakna tampak pada homozygotDijumpai pada orang Eropa Utara,lebih sedikit imsidennya dibanding dengan defisiensi G6PDDefisinei bukan secara kuantitatif,lebih sering menunjukan varian PK dengan karaktertistik abnormal

PatogenisisKelainan glikolisis eritrosit dengan pembentukan ATP berkurang eritrosit menjadi kaku, mengalami deformitas dan rentan secarametabolik maupun fisikGambaran hematologi: hemolisis dengan reticulositosis yang menyolok,poikilositosis dan banyak eritrositmyangmengalami distrosi

Defek Hb
Ggn sintesa hemoglobin: a. sel sabit: HbS tidak larut dan membentuk kristalpada O2 tekanan rendah,sdm bentuk sabit, ggn substitusi aa. Valin yg seharusnya glutamatb. HbC:kristal rhomboid
c. Hb SC, Hb D&E, Hb MGgn sintesa rantai globin: thalasemia

Hemoglobin SDiturunkan sebagai aotuso dominan tidak lengkapHomozygot tidak mensintesis HbA dan eritrosit mengandung 90-100% HbSHeterozygot mempunyai 20-40% HbSTerutama dijumpai pada populasi Negro, carier tertinggi di Afrika Tengah; 20%

HematologiAnemia normokrom normositik sedang sp beratRetikulositosis, neutrofilia sedangTrombosit sering meningkatDarah tepi; polikromasi bertambah,eritrosit berinti (+), sel sabit (+), target sel dan Howell Yolly bodiesLED rendah, sel sabit sulit membentuk rouleauxElektroforesis Hb, HbS bermigrasi > lambat dp HbA

Gambaran klinisBiasanya manifes pada bayi dgn tanda2 fisik kurang berkembang serng menonjolGejala dan tanda2 anemia hemolitikTanda yg berkaitan dgn fenomena trombotik; nyeri abdomen akibat infarck limfa, nyeri dada, nyeri tulang, hematuria ggn neurologis

THALASSEMIAThalassaemia or "thalassemia" is an inherited autosomal recessive blood diseaseTerganggunya salah satu rantai globin yang dapat menyebabkan terbentuknya Hb yang abnornalThalassemia biasanya pada bangsa Mediterranea, Arab, dan Asia

PATOFISIOLOGI THALASSEMIANormal haemoglobin terdiri dari 2 rantai α dan β globinPenderita Thalassemia mengalami gangguan dalam pembentukan α atau β globinThalassemia dibagi menjadi 2 tergentung rantai globin mana yang terganggu α thalassemia, gangguan pada rantai globin α sedang β thalassemia gangguan pada produksi rantai globin β

HEMOGLOBIN NORMAL

- THALASSEMIAMeliputi HBA1 and HBA2α thalassemia disebabkan penurunan produksi apha-globin , rantai alpha-globin memendek,terjadi pemendekan rantai β pada orang dewasa dan pengurangan rantai y pada bayi baru lahir newbornsPengurangan rantai β membentuk “ unstable tetramers” ( Hemoglobin H / HbH dengan 4 rantai beta ) dimana curve disosiasi oxygennya abnormal

HbH
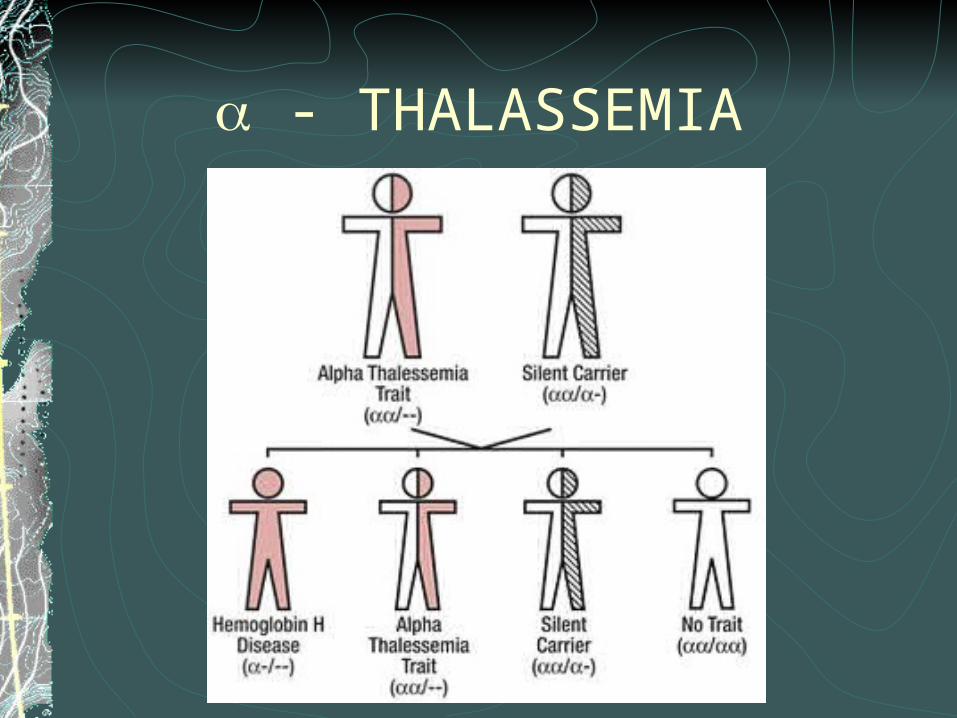
- THALASSEMIA

- THALASSEMIABeta thalassemia karena mutasi HBB gene pada chromosome 11 Berat ringannya penyakit tergantung terjadinya mutasiMutasi khas ( pada βo atauor β thalassemia major) terjadi penekanan pada pembentukan rantai β ( bentuk beta Thalassemia yang paling berat)


GDT THALASSEMIA

ThalasemiaThalasemia β mayor= Cooley3-6 bln An berat, ggn globin rantai beta, rantai alfa mengendap menimbulkan hemolisis.Hati&Limpa >>, hiperplasi ss tl Anemia mikrositik hipokromik, elektroforesis HbF dan HbA-Thalasemia β minor: Biasanya tanpa gejala, anemia mikrositik hipokromik. HbA2 , Fe N,

Thalasemia alfa
Rantai alfa penting utk HbFKehilangan 4 gen alfa : kematian intra uteriKehilangan 3 gen alfa : anemia hemolitik berat: mikrositik hipokromik dg SplenomegaliElektroforesa

Target sel, tear drop sel eliptosit, sperosit, mikrosit

Poikilositosis : target sel, tear drop sel, schistosit, sperosit, hipokrom pd thalas mayor

Anemia sel sabit : sel sabit dan target sel

Wajah thalas Beta mayor : tengkorak menonjol, tulang frontal parietal menonjol.
Maxilla besar

ANEMIA APLASTIKA
Definisi ;anemia yang disebabkan jaringan hemopoetik berkurang, bukan akibat fibrosis atau infiltrasi maligna dari sumsum tulang dan menimbulkan sitopeni dalam darah perifer

Ada 2 tipe :Reversibel; hipoplasia dapat diduga. Biasanya akibat radiasi atau theraphi sitotoksik. Biasanya menyerang sel –sel hemopoetik maturyg cepat membelah dari pluri poten stel selIreversibel; idiopatik primer/ sekunder thd reaksi idiosinkrasi obat atau infeksi virus, mungkin tejadi kelainan/ kerusakan hemopoetik stem sel
Kadang bersifat autoimunTerjadi penurunan jumlah/ kapasitas fungsional sel2 yg bersifat kronik

PATOGENISISBerkuangnya jar hemopoetik dan bertanbahnya jar lemak dalam su-tulProduksi sel ber(-) diawali jumlahgranulosit dan trombositKhas , terjadi pansitopenia

HEMATOLOGIAnemia normokrom normositikReticulopeniaLekopenia, granulosit sering < 1,5 x 10 .000/ml ,matur kadang dgn granulasi toksik. Limfopeni denga sel T relatf/ mutlak ber(-) TrombositopeniHbf meningkat terutama pd anak2S
• SU_TUL : hiposelluler, cadangan besi
